











-
Since the discovery of graphene, researchers have been being increasingly attracted by the emerging of a bunch of two-dimensional (2D) materials, such as BN, MoS2 and black phosphorene. These materials possess outstanding physical and chemical properties, which could find great potential applications in nanoelectronics, energy conversion or storage, photocatalysts, etc. Recently, a theoretically predicted pucker layered material consisting of Sb atoms, antimonene, has been prepared, and is attracting the attention in the field of lithium ion batteries. In this paper, based on first-principle density functional theory, the adsorption characteristics of Li atoms on antimony are studied, including the most stable adsorption configuration, the adsorption density and the diffusion path of Li atom on antimonene. The results show that the most stable adsorption configuration of Li atom is in the valley site, i.e. the center of the three Sb atoms in the top layer and one Sb in the bottom layer. The adsorption energy is 1.69 eV and the adsorption distance is 2.81 Å. The band structure shows that antimony is an indirect band gap semiconductor with a band gap of 1.08 eV. After the absorption of Li atom, the Fermi level enters into the conduction band, which shows an electronic property similar to metal. The analysis of density of states shows that the p-electronic state of Sb atom and the p and s electronic state of Li atom possess distinct resonance peaks, showing hybrid bonding characteristics. With the increase of the number of Li atoms adsorbed, the lattice structure and electronic structure of antimonene change greatly. The nudged elastic band calculation shows that the diffusion barrier of Li atom on antimony surface is 0.07 eV, and a smaller barrier height is beneficial to the rapid charge-discharge process. To sum up, antimony has a good potential as an anode material for lithium ion batteries. -
Keywords:
- antimonene /
- two-dimensional materials /
- lithium atom adsorption /
- density functional theory
[1] Tarascon J M, Armand M 2001 Nature 414 6861
[2] Li Y, Mi Y, Sun G 2015 J. Mater. Chem. 03 12
[3] Geim A K 2009 Science 324 5934
[4] Geim A K, Novoselov K S 2007 Nature Mat. 6 3
 Google Scholar
Google Scholar
[5] Yao Q, Huang C, Yuan Y 2015 J. Phys. Chem. C 119 12
[6] Hussain A, Ullah S, Farhan M A 2017 J. Mater. Chem. 41 19
[7] 孙建平, 周科良, 梁晓东 2016 65 018201
Sun J P, Zhou K L, Liang X D 2016 Acta Phys. Sin. 65 018201
[8] 孙建平, 缪应蒙, 曹相春 2013 62 036301
Sun J P, Miu Y M, Cao X C 2013 Acta Phys. Sin. 62 036301
[9] 高云雷, 赵东林, 白利忠, 张霁明, 孔莹 2012 中国科技论文 7 413
Gao Y L, Zhao D L, Bai L Z, Zhang J M, Kong Y 2012 China Sciencepaper 7 413
[10] 朱晋潇, 刘晓东, 薛敏钊, 陈长鑫 2017 物理化学学报 33 2153
Google Scholar
Zhu J X, Liu X D, Xue M Z, Chen C X 2017 Acta Physico-Chimica Sinica 33 2153
Google Scholar
[11] Peng B, Xu Y L, Fokko M M 2017 Acta Physico-Chimica Sinica 33 2127
[12] Li L K, Yu Y J, Ye G J, Ge Q Q, Ou X D, Wu H, Feng D L, Chen X H, Zhang Y B 2014 J. Nat. Nano 9 372
Google Scholar
[13] Liu H, Neal A T, Zhu Z, Luo Z, Xu X F, Tomanek D, Ye P D 2017 ACS Nano 8 4033
[14] Yao Q, Huang C, Yuan Y 2015 J. Phys. Chem. C 119 12
[15] Qiao J S, Kong X H, Hu Z X, Yang F, Ji W 2014 Nat. Com. 5 128
[16] He Y, Xia F, Shao Z 2015 J. Phys. Chem. C. Lett. 6 23
[17] Haldar S, Mukherjee S, Ahmed F 2017 J. Hyd. Ene. 42 36
[18] Wang G, Pandey R, Karna S P 2015 ACS Appl. Mat. Int. 6 21
[19] Zhang S L, Yan Z, Li Y F, Chen Z F, Zeng H B 2015 Ang. Chem. Int. Edi. 54 3112
Google Scholar
[20] Ji J P, Song X F, Liu J Z, Yan Z, Huo C X , Zhang S L, Su M, Liao L, Wang W H, Ni Z H, Hao Y F, Zeng H B 2016 Nat. Com. 6 133
[21] Ares P, Aguilar-Galindo F, Rodriguez-San-Miguel D, Aldave D A, Diaz-Tendero S, Alcami M, Martin F, Gomez-Herrero J, Zamora F 2016 Adv. Mat. 28 30
[22] Gibaja C, Rodriguez-San-Miguel D, Ares P 2016 Ang. Chem. 55 46
[23] Üzengi Aktürk O, Aktürk E, Ciraci S 2016 Phys. Rev. 93 3
[24] Zhao M W, Zhang X M, Li L Y 2015 Sci. Rep. 5 161
[25] Zhou Y G, Lin X D 2018 Appl. Sur. Sci. 458 572
Google Scholar
[26] Xie M, Zhang S, Cai B 2016 RSC Adv. 6 18
Google Scholar
-
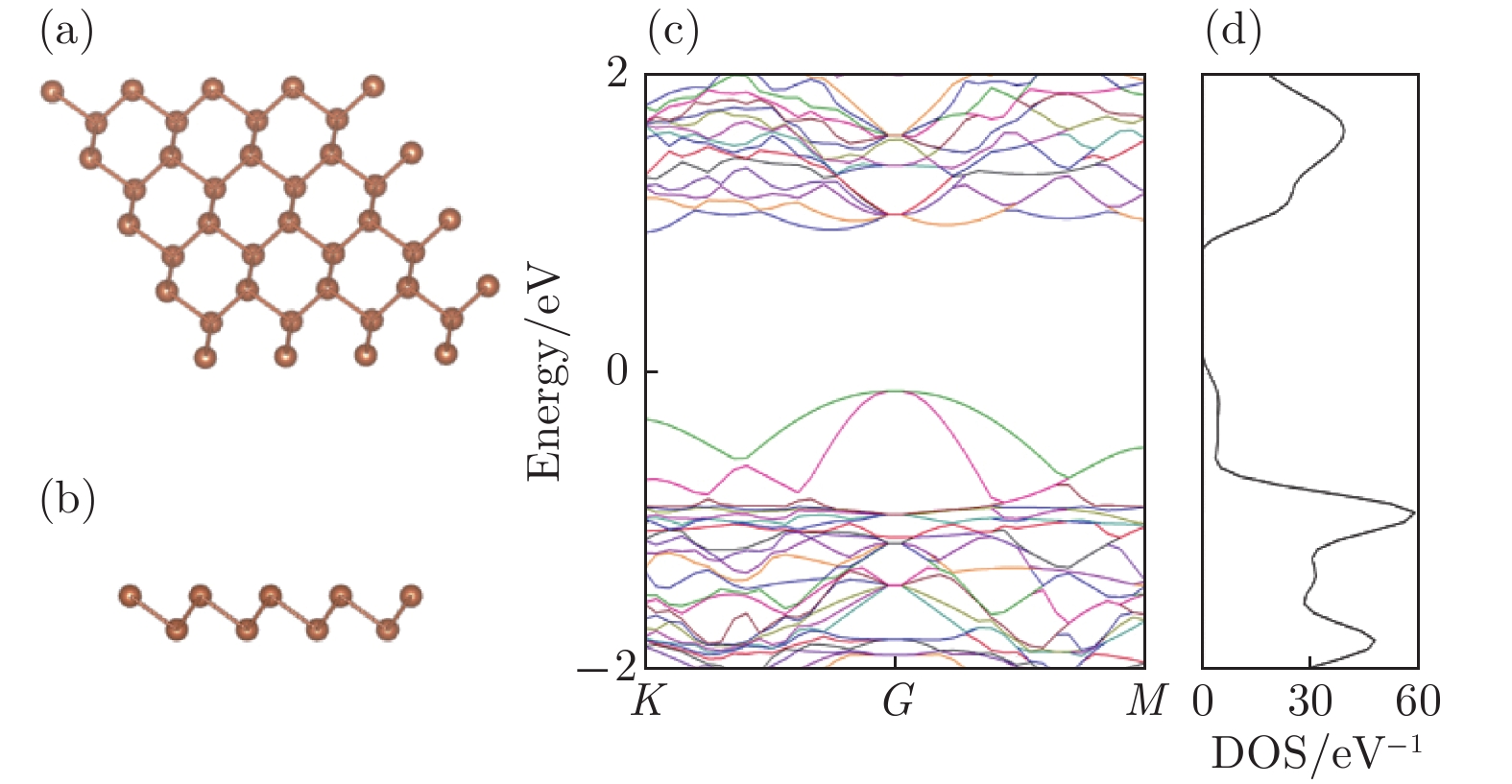
图 1 本征锑烯 (a) 俯视图; (b) 侧视图; (c) 能带图; (d) 态密度图
Figure 1. Pristine antimonene: (a) The top view; (b) the side view; (c) the band structure; (d) the density of states.
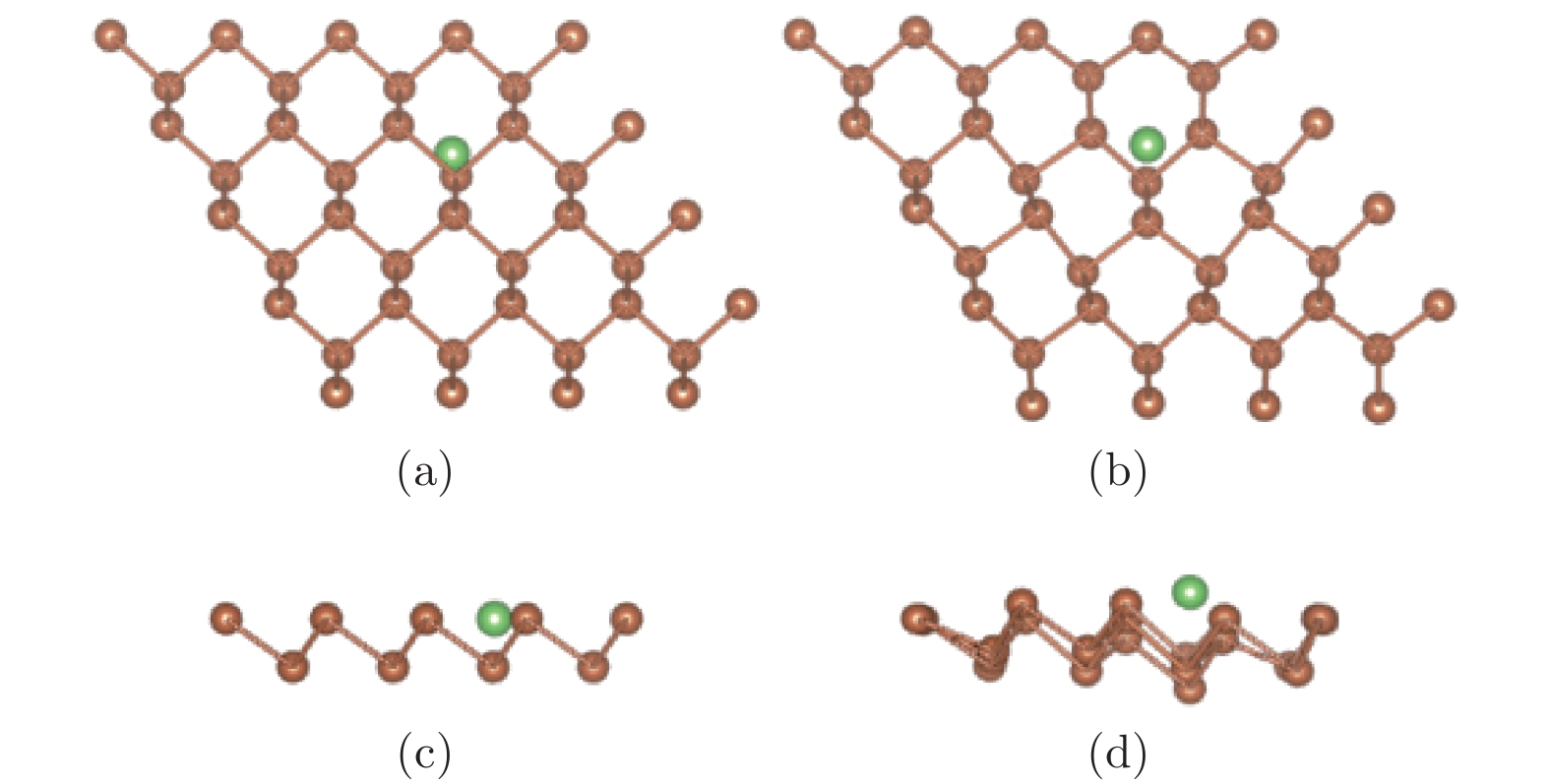
图 2 (a)和(c)分别为谷位吸附结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图
Figure 2. The antimonene structure of Li adsorbed on vacancy site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
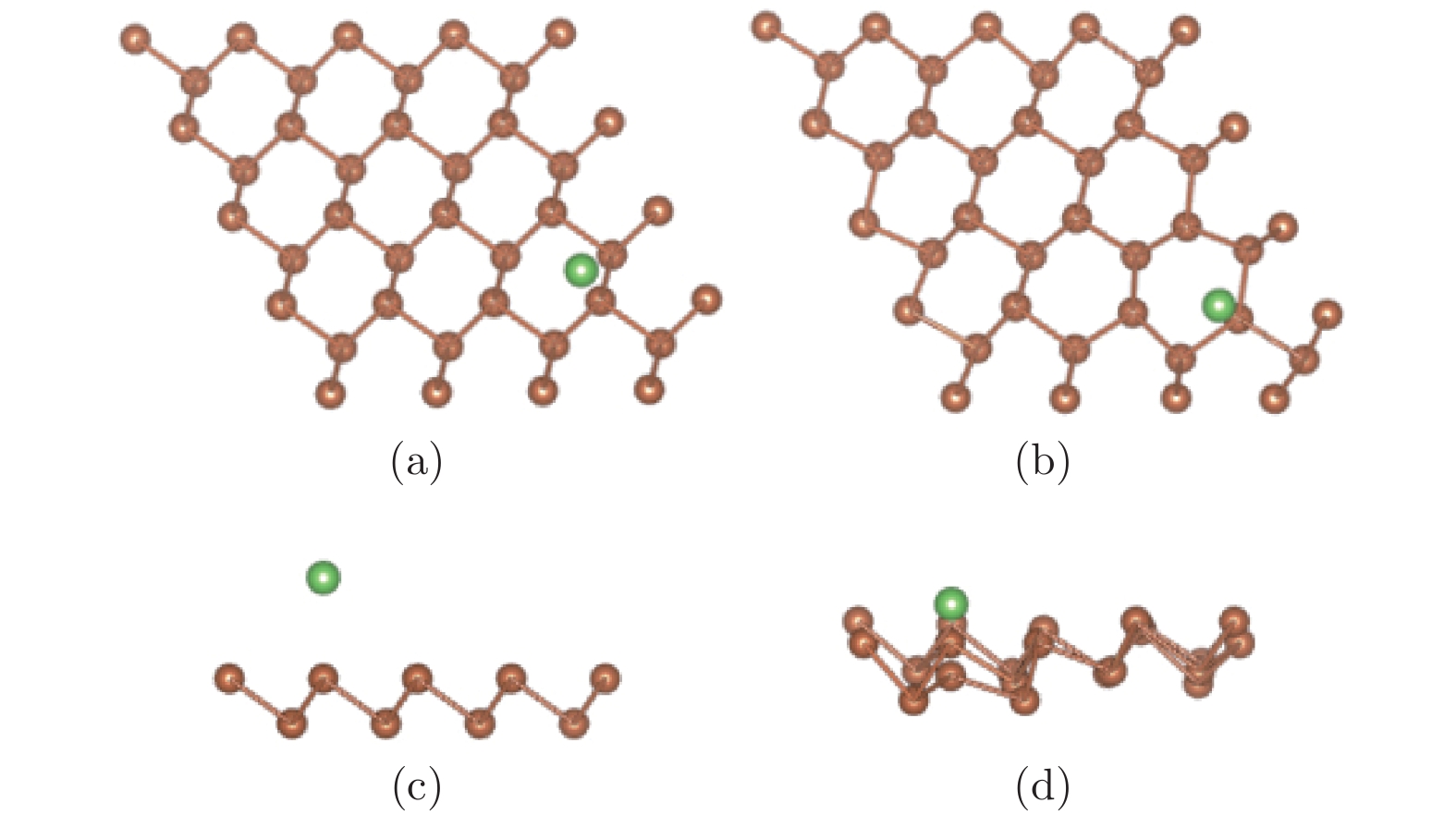
图 3 (a) 和 (c) 分别为顶位吸附结构初始构型的俯视图和侧视图; (b) 和 (d) 分别为其优化构型的俯视图和侧视图
Figure 3. The antimonene structure of Li adsorbed on top site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
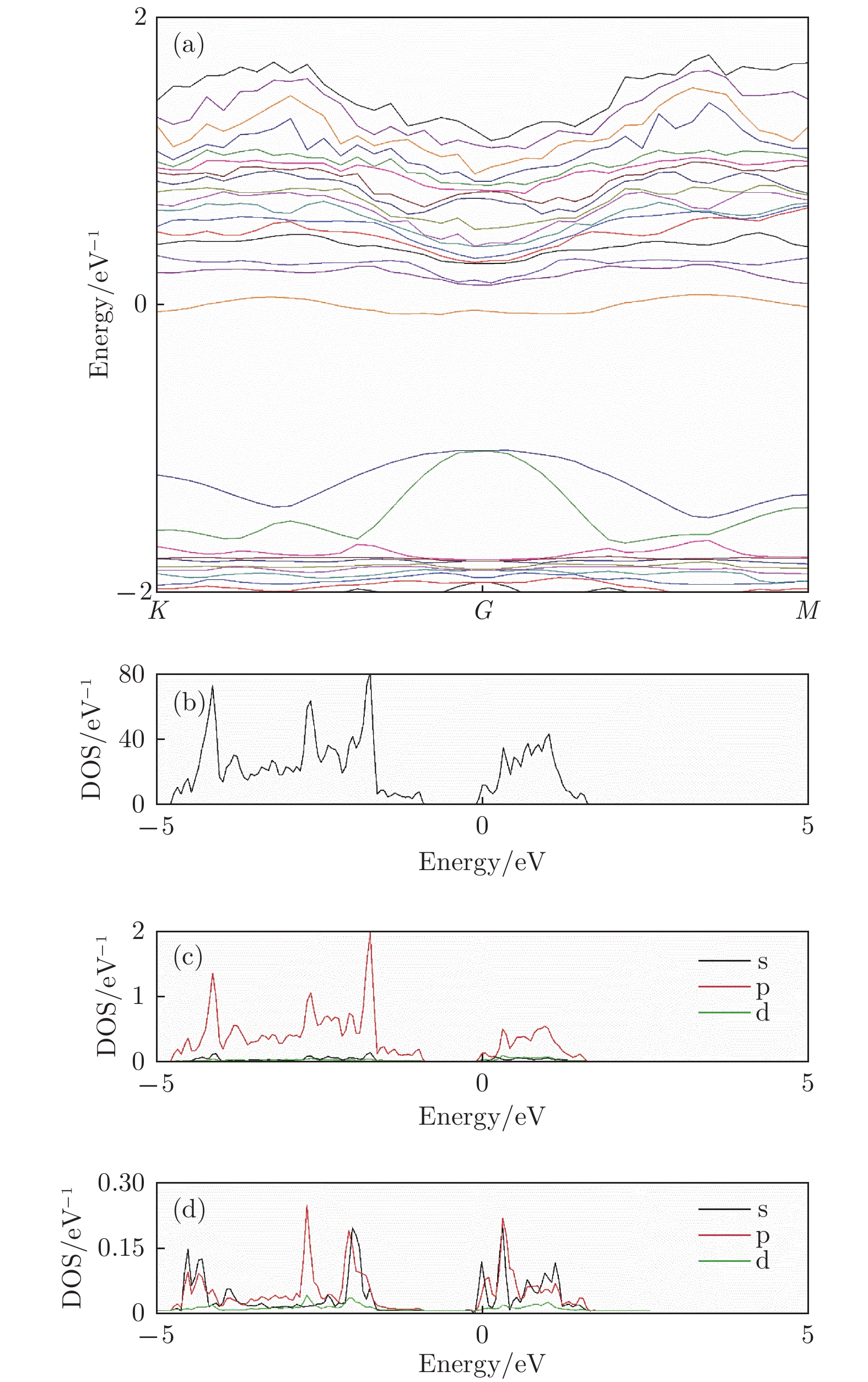
图 4 谷位吸附构型 (a)能带图; (b)总态密度; (c) Sb分波态密度; (d) Li分波态密度
Figure 4. The band structure and density of Li adsorbed antimonene: (a) The band structure; (b) the total density of states; (c) the partial density of states of Sb atom; (d) the partial density of states view of Li atom.
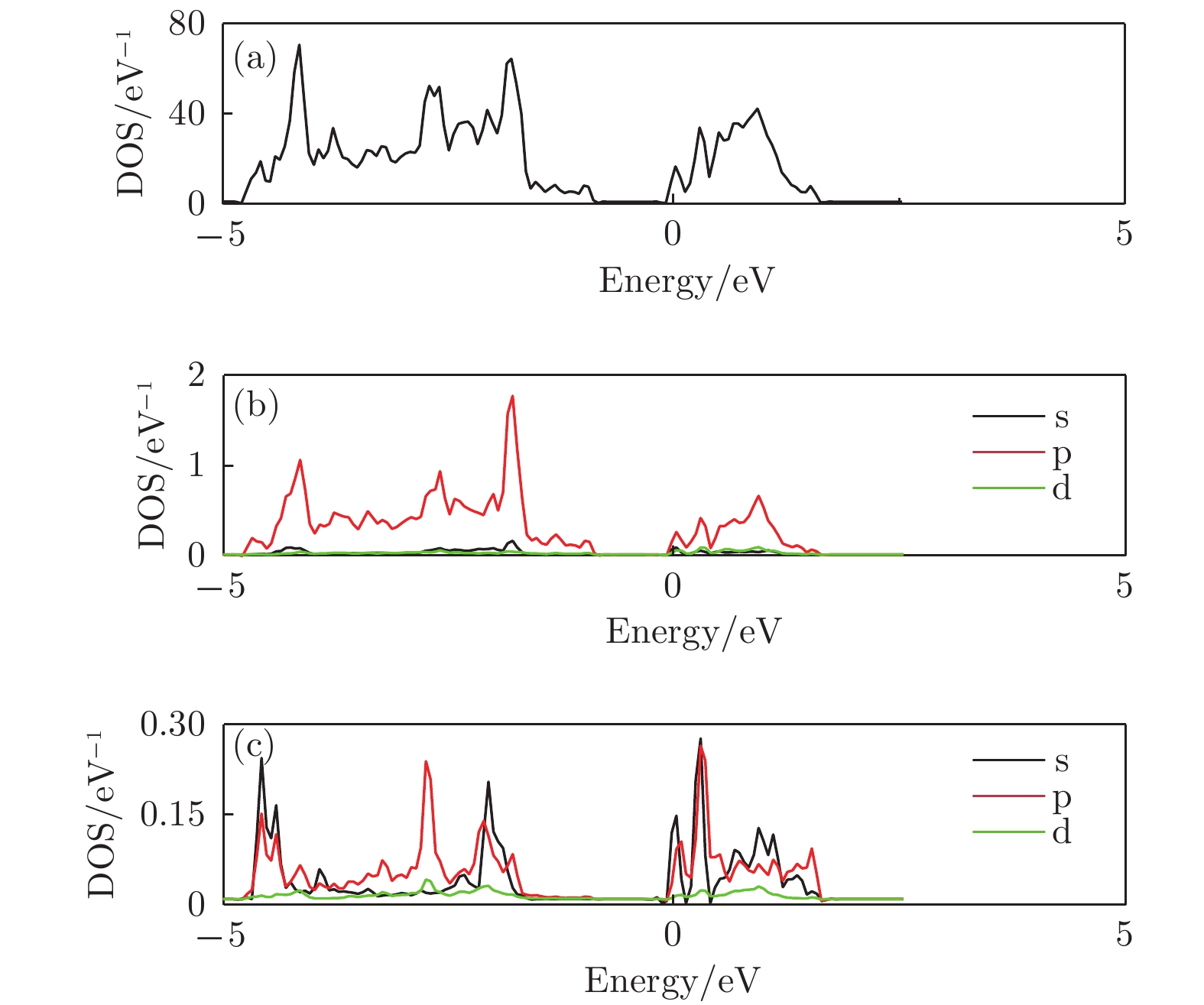
图 5 顶位吸附结构态密度图 (a)总态密度; (b) Sb分波态密度; (c) Li分波态密度
Figure 5. The density of states of the Li adsorbed structure: (a) The total density of states; (b) the partial density of states of Sb atom; (c) the partial density of states view of Li atom.
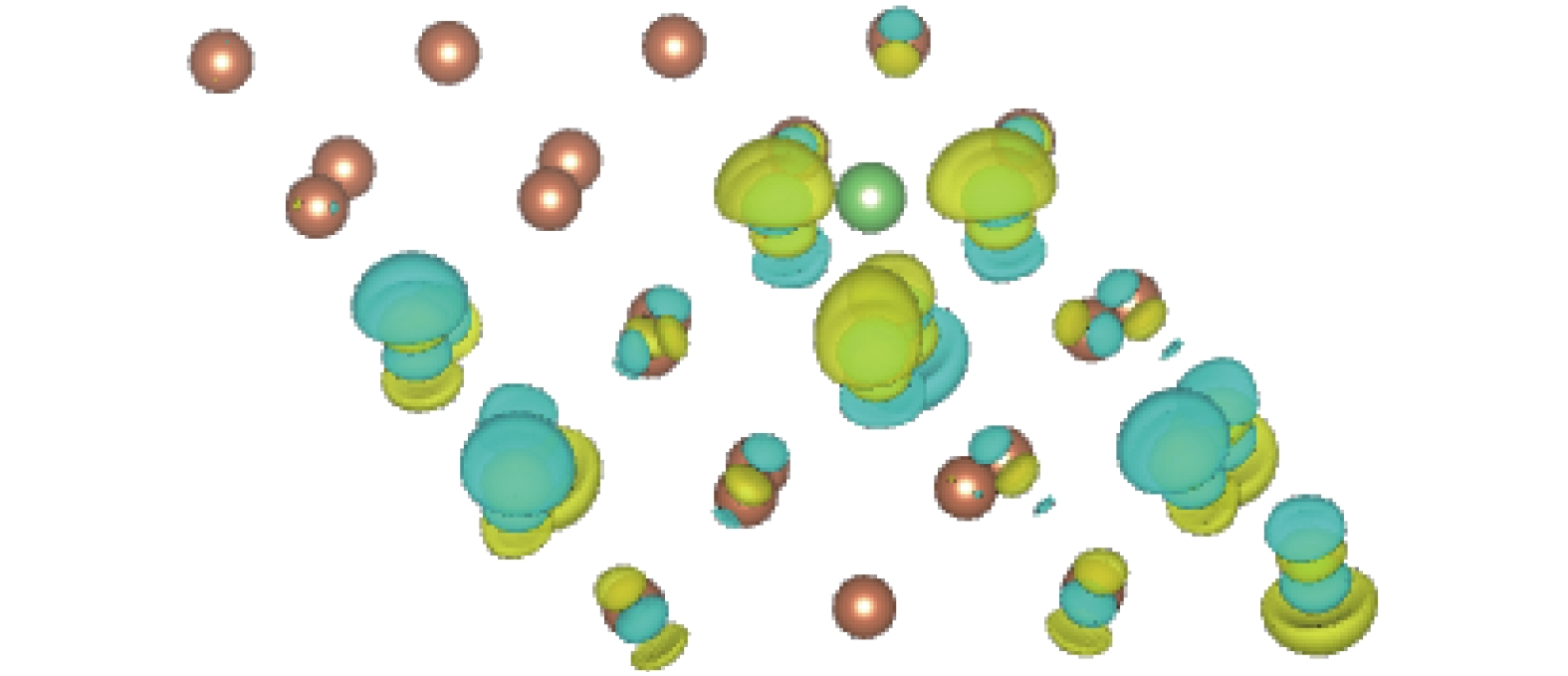
图 6 谷位差分电荷密度
Figure 6. The differential charge density of the Li adsorbed structure.
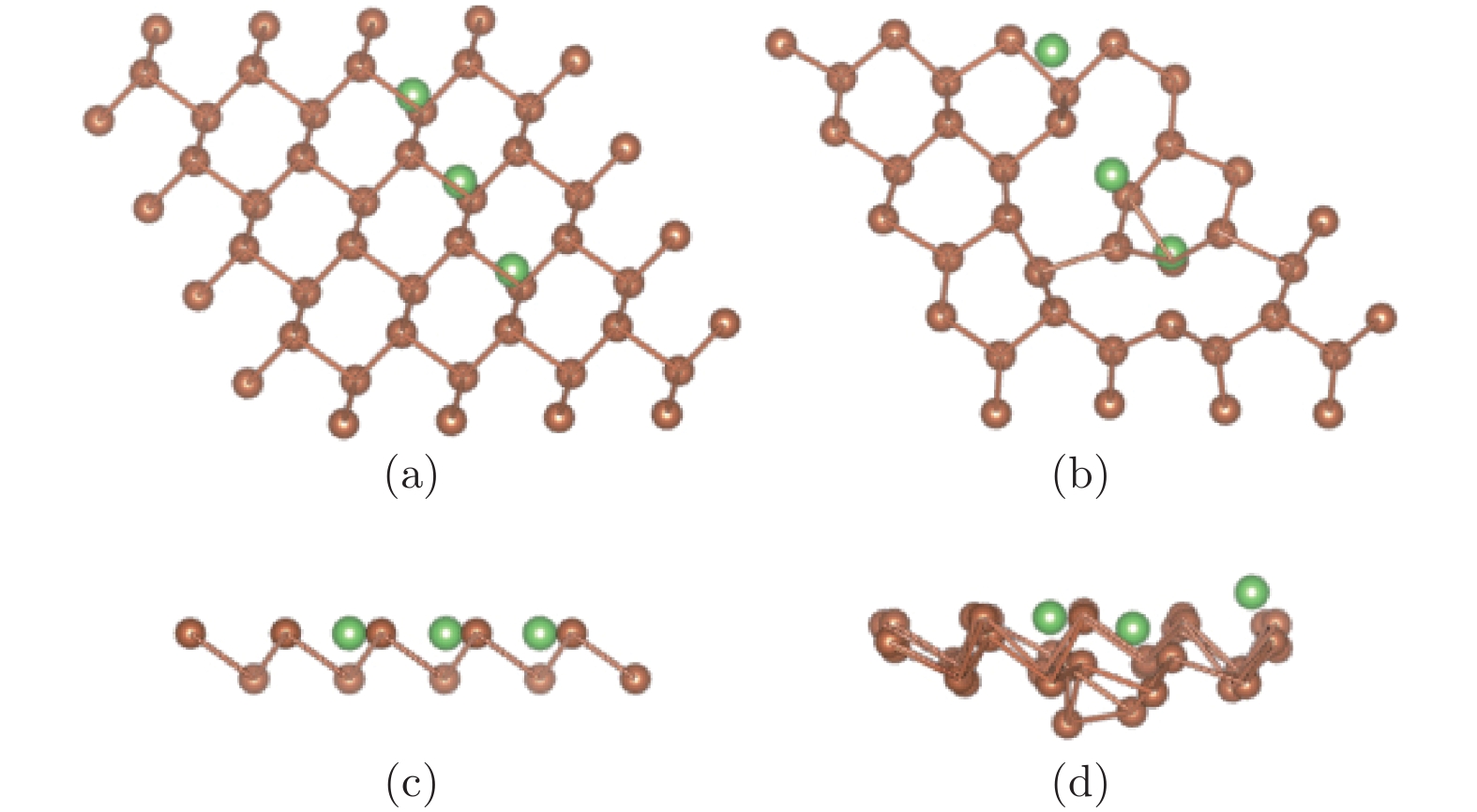
图 7 (a)和(c)分别为锑烯吸附3个Li原子结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图
Figure 7. The configurations of antimonene adsorbing three lithium atoms: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
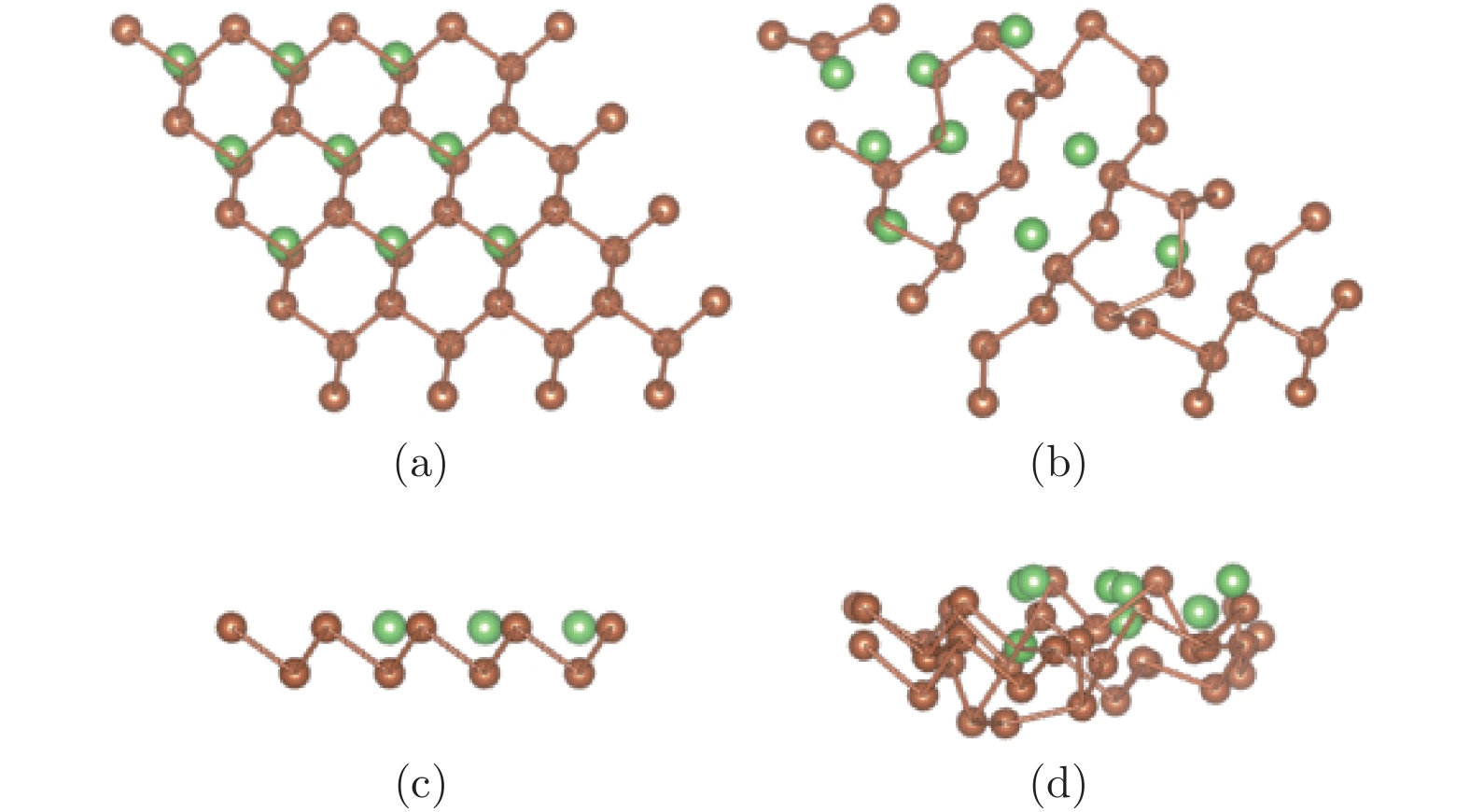
图 8 (a)和(c)分别为锑烯吸附九个Li原子结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图
Figure 8. The configurations of antimonene adsorbing nine lithium atoms: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
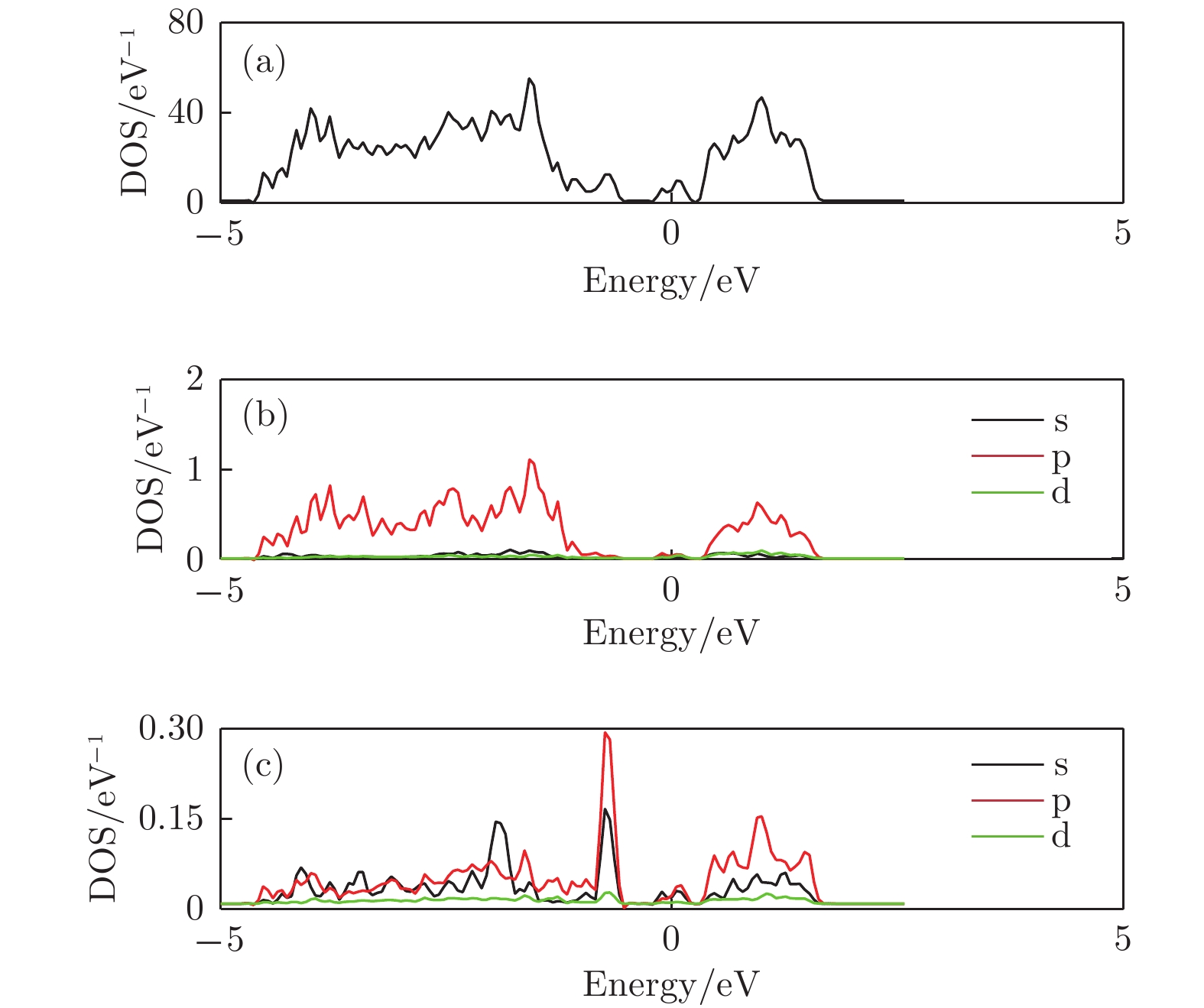
图 9 锑烯吸附三个Li原子态密度图 (a) 吸附结构的总态密度图; (b) Sb原子的分波态密度; (c) Li原子的分波态密度
Figure 9. The density of states of antimonene adsorbing three lithium atoms: (a) The total density of states of the structure; (b) the partial density of states of Sb atom; (c) the partial density of states view of Li atom.
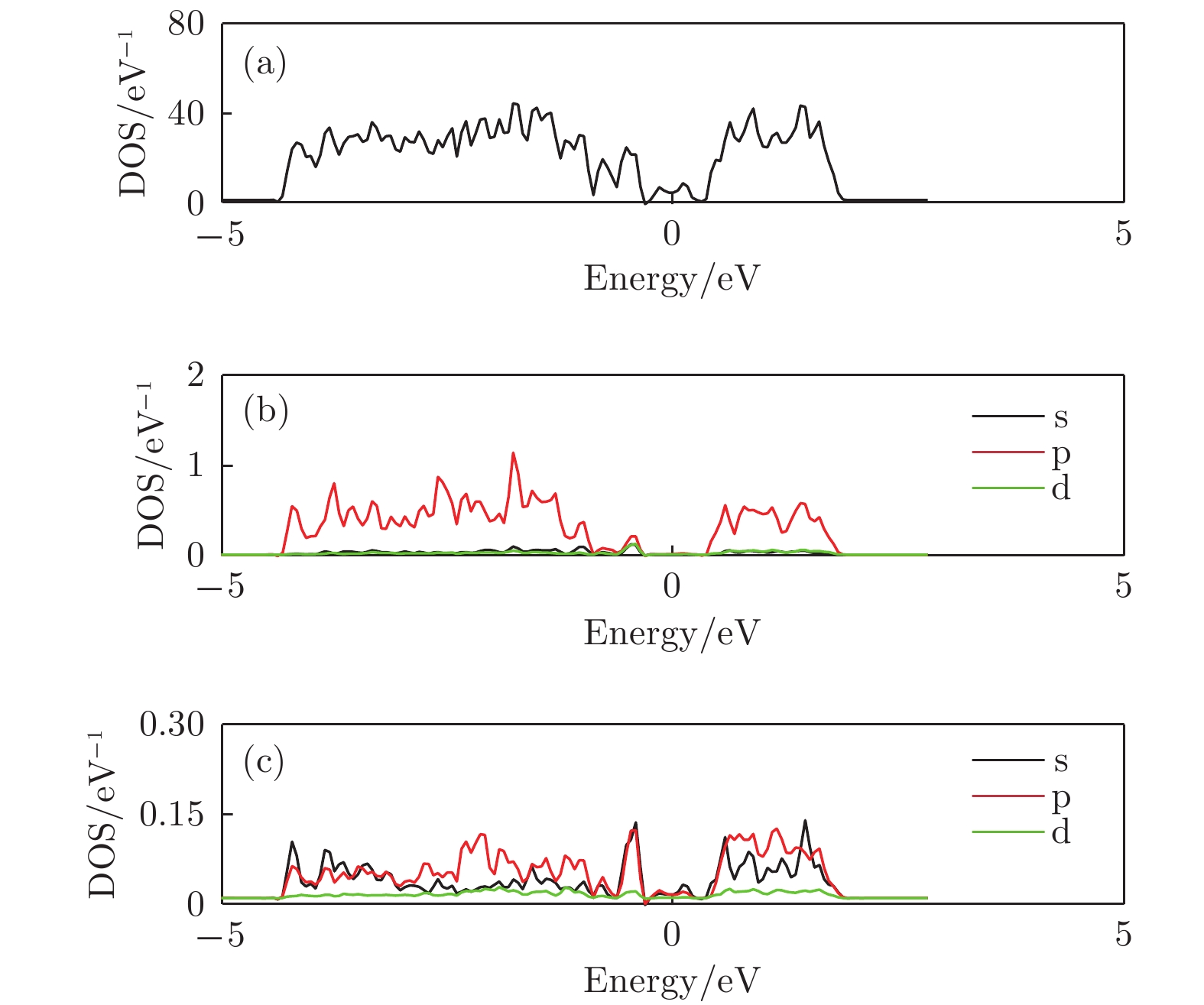
图 10 锑烯吸附九个Li原子态密度图 (a) 吸附体系的总态密度图; (b) 体系中Sb原子的分波态密度; (c) 体系中Li原子的分波态密度
Figure 10. The density of states of antimonene adsorbing nine lithium atoms: (a) The total density of states of the structure ; (b) the partial density of states of Sb atom; (c) the partial density of states view of Li atom.
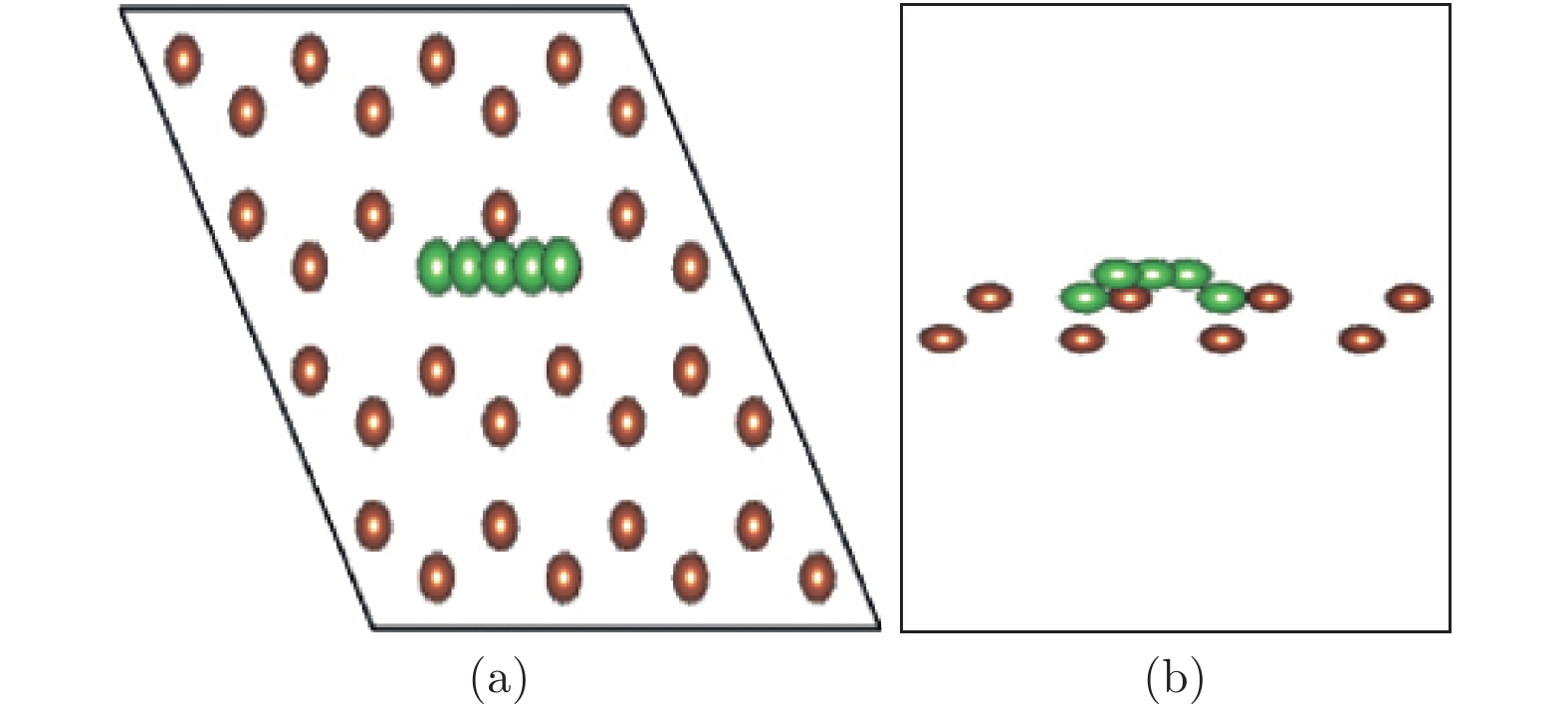
图 11 Li原子扩散迁移路径 (a) 俯视图; (b) 侧视图
Figure 11. The Li diffusion path on antimonene: (a) The top view; (b) the side view.
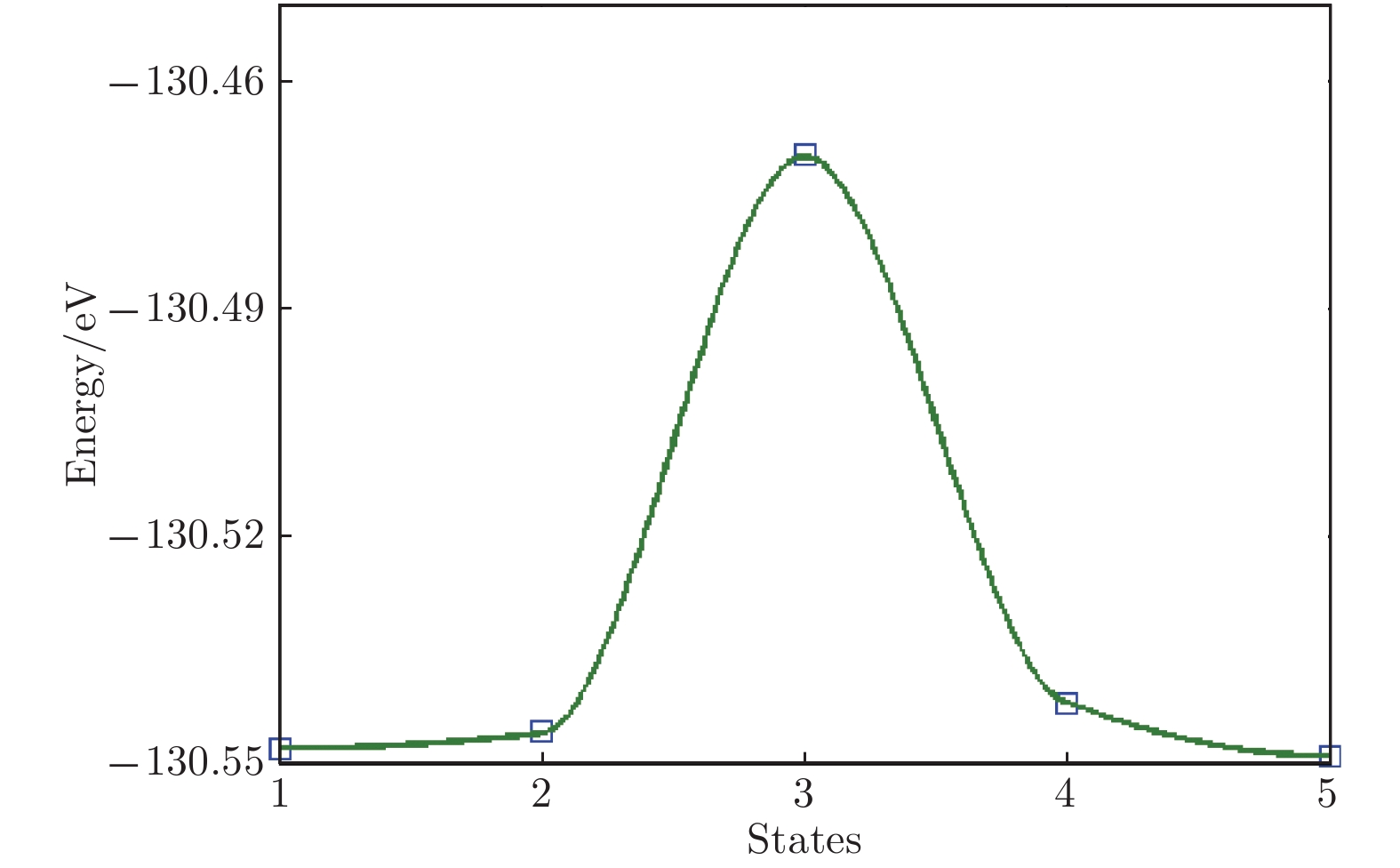
图 12 Li原子扩散迁移势垒
Figure 12. The diffusion energy barrier of Li atom.
表 1 锑烯吸附Li原子的两种结构的特性
Table 1. Properties of two Li adsorbed antimonene configurations.
Ead/eV Sb—Li/Å Barder(Sb)/Δq Barder(Li)/Δq 顶位 1.67 2.83 0.31 −1.00 谷位 1.69 2.81 0.32 −0.99 注: Ead表示吸附能, Sb—Li表示在吸附后的Sb—Li键长.  DownLoad: CSV
DownLoad: CSV
-
[1] Tarascon J M, Armand M 2001 Nature 414 6861
[2] Li Y, Mi Y, Sun G 2015 J. Mater. Chem. 03 12
[3] Geim A K 2009 Science 324 5934
[4] Geim A K, Novoselov K S 2007 Nature Mat. 6 3
Google Scholar
[5] Yao Q, Huang C, Yuan Y 2015 J. Phys. Chem. C 119 12
[6] Hussain A, Ullah S, Farhan M A 2017 J. Mater. Chem. 41 19
[7] 孙建平, 周科良, 梁晓东 2016 65 018201
Sun J P, Zhou K L, Liang X D 2016 Acta Phys. Sin. 65 018201
[8] 孙建平, 缪应蒙, 曹相春 2013 62 036301
Sun J P, Miu Y M, Cao X C 2013 Acta Phys. Sin. 62 036301
[9] 高云雷, 赵东林, 白利忠, 张霁明, 孔莹 2012 中国科技论文 7 413
Gao Y L, Zhao D L, Bai L Z, Zhang J M, Kong Y 2012 China Sciencepaper 7 413
[10] 朱晋潇, 刘晓东, 薛敏钊, 陈长鑫 2017 物理化学学报 33 2153
Google Scholar
Zhu J X, Liu X D, Xue M Z, Chen C X 2017 Acta Physico-Chimica Sinica 33 2153
Google Scholar
[11] Peng B, Xu Y L, Fokko M M 2017 Acta Physico-Chimica Sinica 33 2127
[12] Li L K, Yu Y J, Ye G J, Ge Q Q, Ou X D, Wu H, Feng D L, Chen X H, Zhang Y B 2014 J. Nat. Nano 9 372
Google Scholar
[13] Liu H, Neal A T, Zhu Z, Luo Z, Xu X F, Tomanek D, Ye P D 2017 ACS Nano 8 4033
[14] Yao Q, Huang C, Yuan Y 2015 J. Phys. Chem. C 119 12
[15] Qiao J S, Kong X H, Hu Z X, Yang F, Ji W 2014 Nat. Com. 5 128
[16] He Y, Xia F, Shao Z 2015 J. Phys. Chem. C. Lett. 6 23
[17] Haldar S, Mukherjee S, Ahmed F 2017 J. Hyd. Ene. 42 36
[18] Wang G, Pandey R, Karna S P 2015 ACS Appl. Mat. Int. 6 21
[19] Zhang S L, Yan Z, Li Y F, Chen Z F, Zeng H B 2015 Ang. Chem. Int. Edi. 54 3112
Google Scholar
[20] Ji J P, Song X F, Liu J Z, Yan Z, Huo C X , Zhang S L, Su M, Liao L, Wang W H, Ni Z H, Hao Y F, Zeng H B 2016 Nat. Com. 6 133
[21] Ares P, Aguilar-Galindo F, Rodriguez-San-Miguel D, Aldave D A, Diaz-Tendero S, Alcami M, Martin F, Gomez-Herrero J, Zamora F 2016 Adv. Mat. 28 30
[22] Gibaja C, Rodriguez-San-Miguel D, Ares P 2016 Ang. Chem. 55 46
[23] Üzengi Aktürk O, Aktürk E, Ciraci S 2016 Phys. Rev. 93 3
[24] Zhao M W, Zhang X M, Li L Y 2015 Sci. Rep. 5 161
[25] Zhou Y G, Lin X D 2018 Appl. Sur. Sci. 458 572
Google Scholar
[26] Xie M, Zhang S, Cai B 2016 RSC Adv. 6 18
Google Scholar


 DownLoad:
DownLoad:











Catalog
Metrics
- Abstract views: 14171
- PDF Downloads: 158
- Cited By: 0
