










-
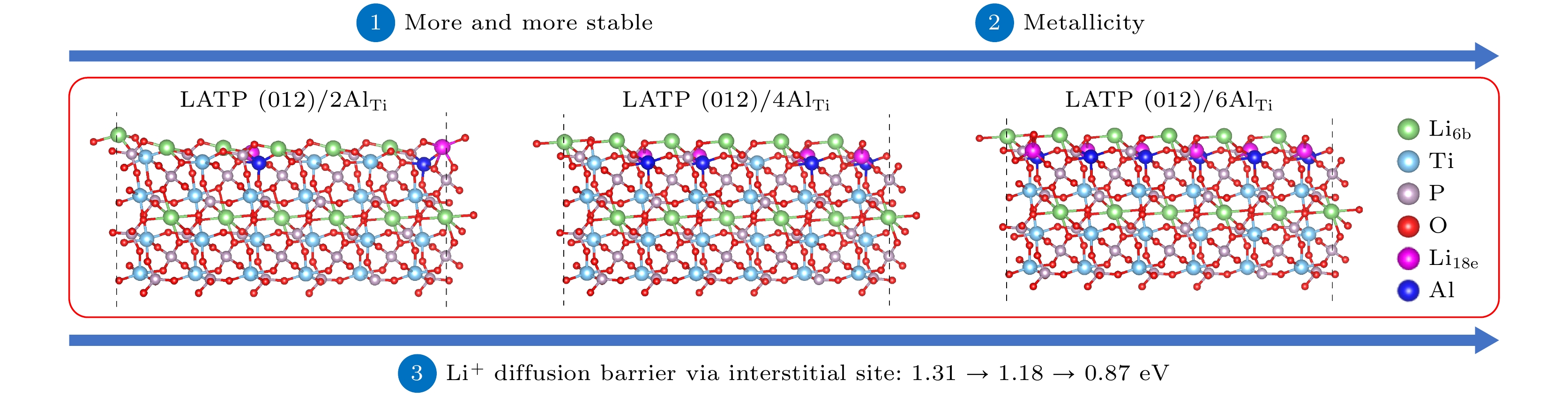
NASICON型Li1+xAlxTi2–x(PO4)3 (LATP)作为锂离子电池极具潜力的固态电解质而备受瞩目. 本文采用第一性原理计算与分子动力学模拟相结合的方法对3种Al掺杂浓度(2AlTi, 4AlTi, 6AlTi)的LATP表面进行研究, 深入探究Al含量对LATP表面的稳定性、电子导电性及Li+输运特性的影响. 研究结果表明, Li-原子终端的(012)面为最稳定晶面, 且LATP(012)表面随Al含量的增大而更为稳定. 电子结构分析表明, LiTi2(PO4)3 (LTP)表面保持与体相一致的半导体性质, 而LATP表面则呈现出金属性, 这是LATP表面锂枝晶生长的一个原因; Li+输运性质的研究结果表明, 对于LTP/LATP表面, 高势垒(大于2.00 eV)使得Li+不能从次表层迁移到最表层; 在最表层中, Li+的最低迁移势垒为0.87 eV, 明显高于其体相的最小值 (0.34 eV). 缓慢的Li+迁移速度是LATP表面锂枝晶生长的另一个重要原因. 幸运的是, 通过提高Al掺杂浓度, 可降低Li+的迁移势垒, 进而提高Li+在LATP表面的扩散性能. 分子动力学模拟进一步揭示Li+在LATP表面的扩散行为主要受到Al含量、Li+占位以及环境温度这些因素的共同影响. 因此, LATP表面的金属性和较高的Li+迁移势垒是其表面锂枝晶生长的两个重要原因. 通过调控Al含量、Li+占位以及环境温度能够不同程度地缓解LATP表面的锂枝晶生长.NASICON-type Li1+xAlxTi2–x(PO4)3 (LATP), as a promising solid-state electrolyte for lithium-ion batteries, has received significant attention due to its simple preparation, low material cost, and good stability in water and air, but the formation of lithium dendrite greatly limits the applications. To elucidate the source of formation of lithium dendrite, in this study, the effects of Al content on the stability, electronic and Li+ mobility properties of the LATP surface with three Al doping concentrations (2AlTi, 4AlTi, 6AlTi) are investigated by combining first-principles calculations and molecular dynamics simulations. The LiTi2(PO4)3 (LTP) surface is also considered for comparison. The results indicate that the (012) surface terminated with Li atoms is the most stable facet. Further, the surface energy of LATP(012) decreases from 0.68 J/m2 to 0.43 J/m2 with the increase of Al content, suggesting that Al doping can effectively improve the stability of the LATP(012) surface. Electronic structure analysis reveals that the surface of LTP(012) retains the semiconductor properties consistent with the bulk phase, whereas the LATP(012) surface exhibits metallicity, which provides an electron pathway for forming the metallic Li . Consequently, the metallic characteristic of the LATP(012) surface is a reason for its lithium dendrite growth. For the Li+ transport properties, two different migration modes: vacancy migration and interstitial migration, are included. When Li+ migrates within the outermost surface, the migration barrier via vacancy is 1.67/1.69 eV for the LTP/LATP (012) surface, while the migration barrier via interstitial is 1.16 eV for LTP(012) and decreases from 1.31 to 0.87 eV with the increase of Al content for LATP(012). Obviously, within the outermost surface, Al doping can reduce the migration barrier of Li+. When Al doping concentration is 6AlTi, the migration barrier is lowest (0.87 eV). Nevertheless, the lowest migration barrier (0.87 eV) for Li+ on the LATP surface is significantly higher than its bulk minimum value of 0.34 eV. When Li+ migrates from the subsurface layer to the outermost surface, the migration barrier is 2.76 eV for LTP(012) and 2.05 eV, 3.20 eV, and 3.06 eV for LATP(012) with 2AlTi, 4AlTi, and 6AlTi content, respectively. All these migration barriers are greater than 2.00 eV, which prevents Li+ from migrating from the subsurface layer to the outermost surface for both LTP and LATP surfaces. Hence, the slow Li+ migration represents another important factor contributing to lithium dendrite growth on the LATP surface. Fortunately, increasing the Al doping concentration can reduce the migration barrier of Li+ and thus enhance its diffusion performance on the LATP surface. Molecular dynamics simulations further reveal that the diffusion behavior of Li+ on the LATP surface is influenced by a combination of factors, including Al content, Li+ occupancy, and ambient temperature. In particular, LATP(012)/6AlTi, LATP(012)/4AlTi, and LATP(012)/2AlTi possess their highest Li+ diffusion coefficients at 900 K, 1100 K, and 1300 K, respectively. Besides, Li+ near the Al doping site is easier to diffuse on the LATP(012) surface. Thus, our study indicates that by changing Al content, Li+ occupation positions, and the temperature, the Li+ diffusion performance of LATP(012) can be effectively modified, thereby suppressing the formation of lithium dendrites on the LATP(012) surface.
-
Keywords:
- lithium-ion batteries /
- solid-state electrolyte /
- LATP surface /
- first-principles calculations /
- Al content
[1] Zhang S, Ma J, Dong S M, Cui G L 2023 Electrochem. Energy Rev. 6 4
 Google Scholar
Google Scholar
[2] Manthiram A, Yu X W, Wang S F 2017 Nat. Rev. Mater. 2 16103
Google Scholar
[3] Bachman J C, Muy S, Grimaud A, Chang H H, Pour N, Lux S F, Paschos O, Maglia F, Lupart S, Lamp P, Giordano L, Shao-Horn Y 2016 Chem. Rev. 116 140
Google Scholar
[4] Fan L, Wei S Y, Li S Y, Li Q, Lu Y Y 2018 Adv. Energy Mater. 8 1702657
Google Scholar
[5] Zhang Z Z, Shao Y J, Lotsch B, Hu Y S, Li H, Janek J, Nazar L F, Nan C W, Maier J, Armand M, Chen L Q 2018 Energy Environ. Sci. 11 1945
Google Scholar
[6] Zheng F, Kotobuki M, Song S F, Lai M O, Lu L 2018 J. Power Sources 389 198
Google Scholar
[7] Subramanian M, Subramanian R, Clearfield A 1986 Solid State Ion. 18&19 562
Google Scholar
[8] Adachi G y, Imanaka N, Aono H 1996 Adv. Mater. 8 127
Google Scholar
[9] Aono H, Sugimoto E, Sadaoka Y, Imanaka N, Adachi G Y 1990 J. Electrochem. Soc. 137 1023
Google Scholar
[10] Schroeder M, Glatthaar S, Binder J R 2011 Solid State Ion. 201 49
Google Scholar
[11] Mariappan C R, Gellert M, Yada C, Rosciano F, Roling B 2012 Electrochem. Commun. 14 25
Google Scholar
[12] Yin F S, Zhang Z J, Fang Y L, Sun C W 2023 J. Energy Storage 73 12
Google Scholar
[13] Arbi K, Lazarraga M G, Chehimi D B, Ayadi-Trabelsi M, Rojo J M, Sanz J 2004 Chem. Mater. 16 255
Google Scholar
[14] Arbi K, Hoelzel M, Kuhn A, García-Alvarado F, Sanz J 2013 Inorg. Chem. 52 9290
Google Scholar
[15] Monchak M, Hupfer T, Senyshyn A, Boysen H, Chernyshov D, Hansen T, Schell K G, Bucharsky E C, Hoffmann M J, Ehrenberg H 2016 Chem. Mater. 55 2941
Google Scholar
[16] Luo Y Y, Liu X Y, Wen C J, Ning T X, Jiang X X, Lu A X 2023 Appl. Phys. A 129 13
Google Scholar
[17] Liang Y J, Peng C, Kamiike Y, Kuroda K, Okido M 2019 J. Alloy. Compd. 775 1147
Google Scholar
[18] Tian H K, Jalem R, Gao B, Yamamoto Y, Muto S, Sakakura M, Iriyama Y, Tateyama Y 2020 ACS Appl. Mater. Interface 12 54752
Google Scholar
[19] Wu P, Zhou W, Su X, Li J, Su M, Zhou X, Sheldon B W, Lu W 2023 Adv. Energy Mater. 13 2203440
Google Scholar
[20] Stegmaier S, Schierholz R, Povstugar I, Barthel J, Rittmeyer S P, Yu S, Wengert S, Rostami S, Kungl H, Reuter K 2021 Adv. Energy Mater. 11 2100707
Google Scholar
[21] Kresse G, Hafner J 1994 J. Phys.: Condens. Matter 6 8245
Google Scholar
[22] Kresse G, Hafner J 1993 Phys. Rev. B 47 558
Google Scholar
[23] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[24] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[25] Perdew J P, Ernzerhof M, Burke K 1996 Chem. Phys. 105 9982
Google Scholar
[26] Pack J D, Monkhorst H J 1977 Phys. Rev. B 16 1748
Google Scholar
[27] Henkelman G, Uberuaga B P, Jónsson H 2000 Chem. Phys. 113 9901
Google Scholar
[28] Nosé S 1984 J. Chem. Phys. 81 511
Google Scholar
[29] Tian H K, Liu Z, Ji Y Z, Chen L Q, Qi Y 2019 Chem. Mater. 31 7351
Google Scholar
[30] 李梅, 钟淑英, 胡军平, 孙宝珍, 徐波 2024 73 362
Google Scholar
Li M, Zhong S Y, Hu J P, Sun B Z, Xu B 2024 Acta Phys. Sin. 73 362
Google Scholar
[31] Han F D, Westover A S, Yue J, Fan X L, Wang F, Chi M F, Leonard D N, Dudney N J, Wang H, Wang C S 2019 Nat. Energy 4 187
Google Scholar
[32] Lang B, Ziebarth B, Elsässer C 2015 Chem. Mater. 27 5040
Google Scholar
[33] Yang K, Chen L K, Ma J B, He Y B, Kang F Y 2021 InfoMat. 3 1195
Google Scholar
-
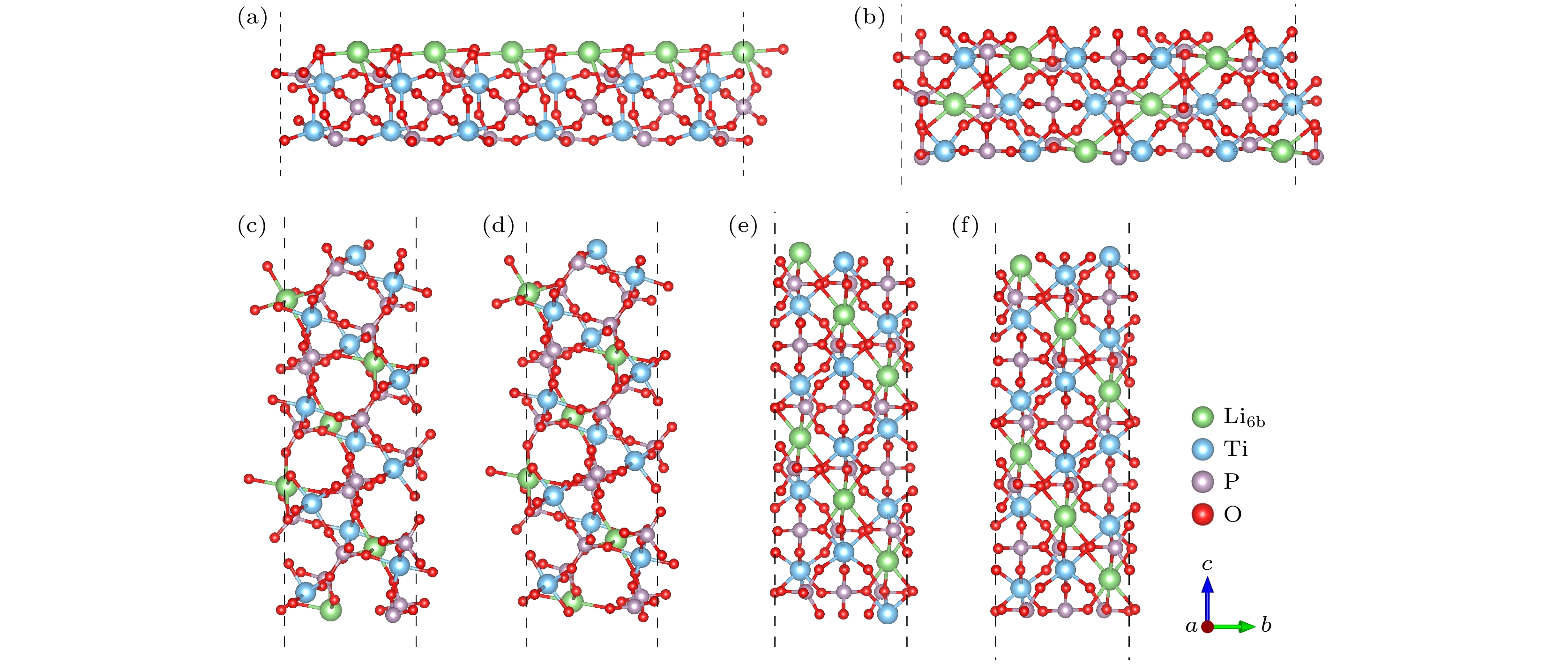
图 1 不同晶面的LTP表面结构图 (a) Li-LTP (012); (b) O-LTP (100); (c) O-LTP (101); (d) Ti-LTP (101); (e) Li-LTP (001); (f) Ti/O-LTP (001)
Fig. 1. LTP surfaces with different crystal face: (a) Li-LTP (012); (b) O-LTP (100); (c) O-LTP (101); (d) Ti-LTP (101); (e) Li-LTP (001); (f) Ti/O-LTP (001).
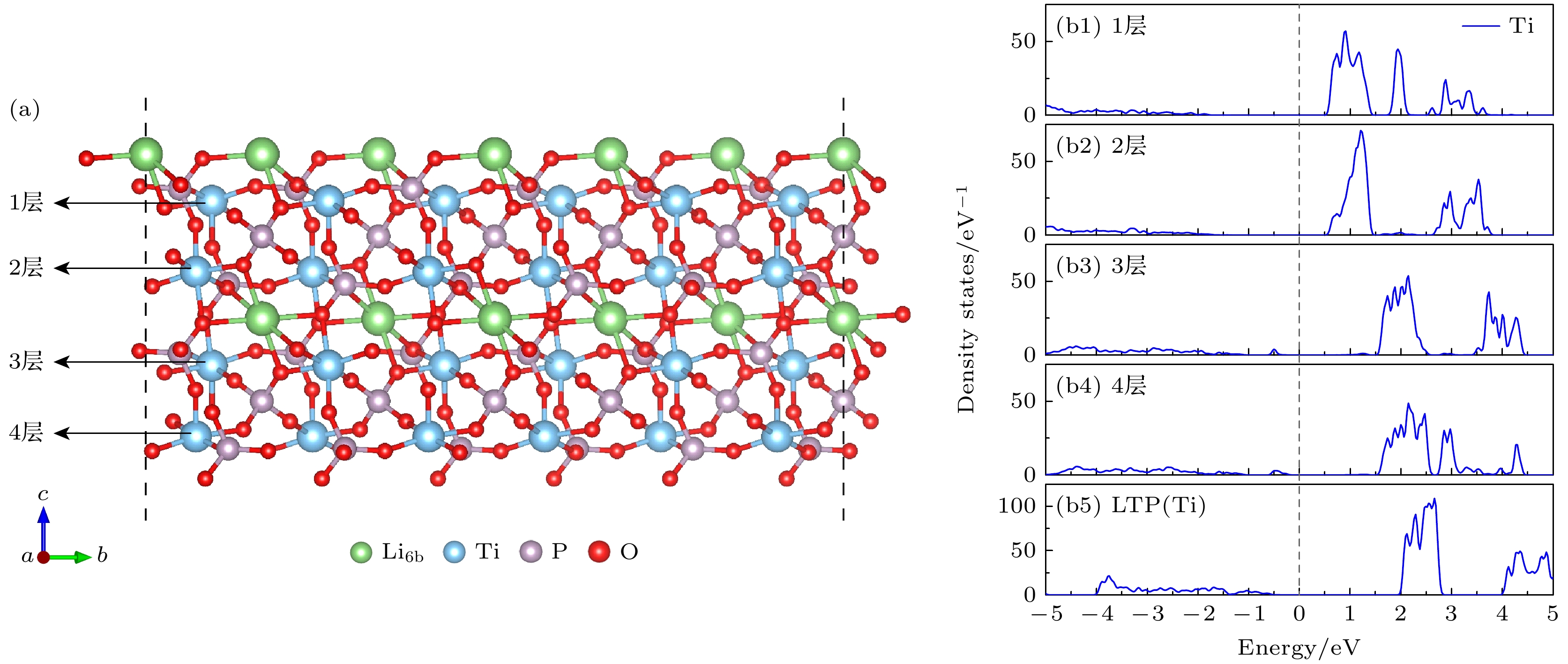
图 2 (a) 含有4层Ti原子层的Li-LTP(012)面的表面结构; (b1)—(b5) Li-LTP(012)表面中每个Ti原子层和LTP体相中Ti的投影态密度图, 能量为0处设为费米能级
Fig. 2. (a) The atomic structure of the Li-LTP(012) surface with 4 Ti atomic layers; (b1)–(b5) the partial density of states (PDOS) corresponding to each Ti atomic layer on the Li-LTP(012) surface and the Ti layers in the LTP bulk, the energy level at 0 is set as the Fermi level.
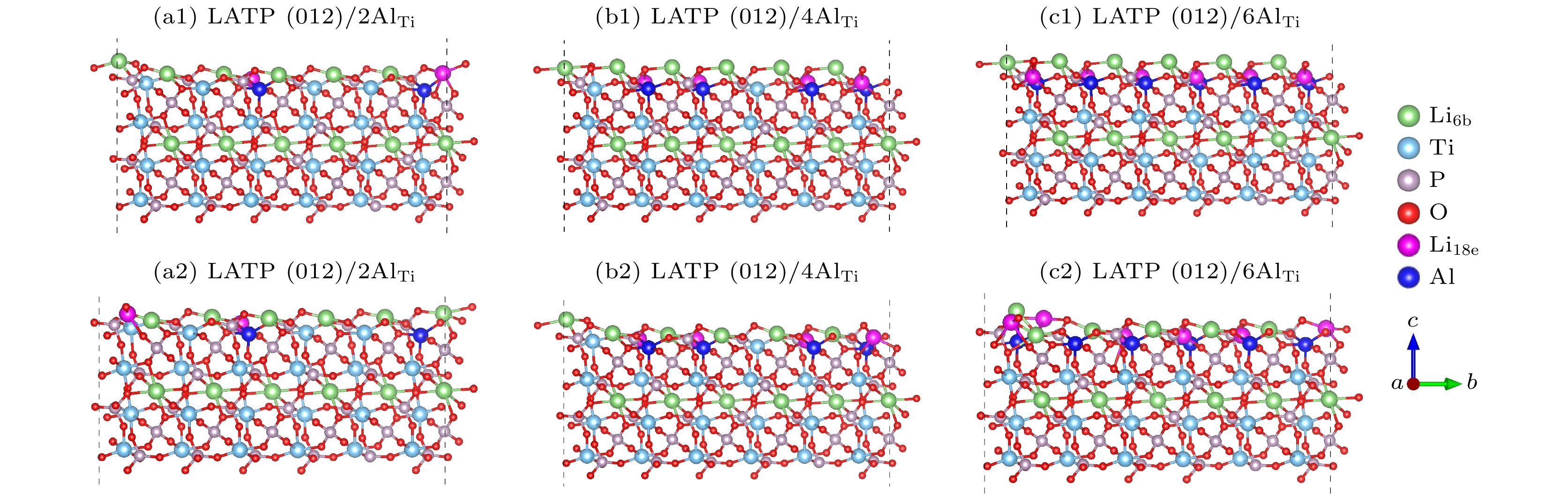
图 3 (a1)—(c1) 优化前不同Al含量下的LATP(012)表面模型; (a2)—(c2) 优化后不同Al含量下的LATP(012)表面模型
Fig. 3. LATP(012) surface with different Al contents before optimization (a1)–(c1) and after optimization (a2)–(c2).
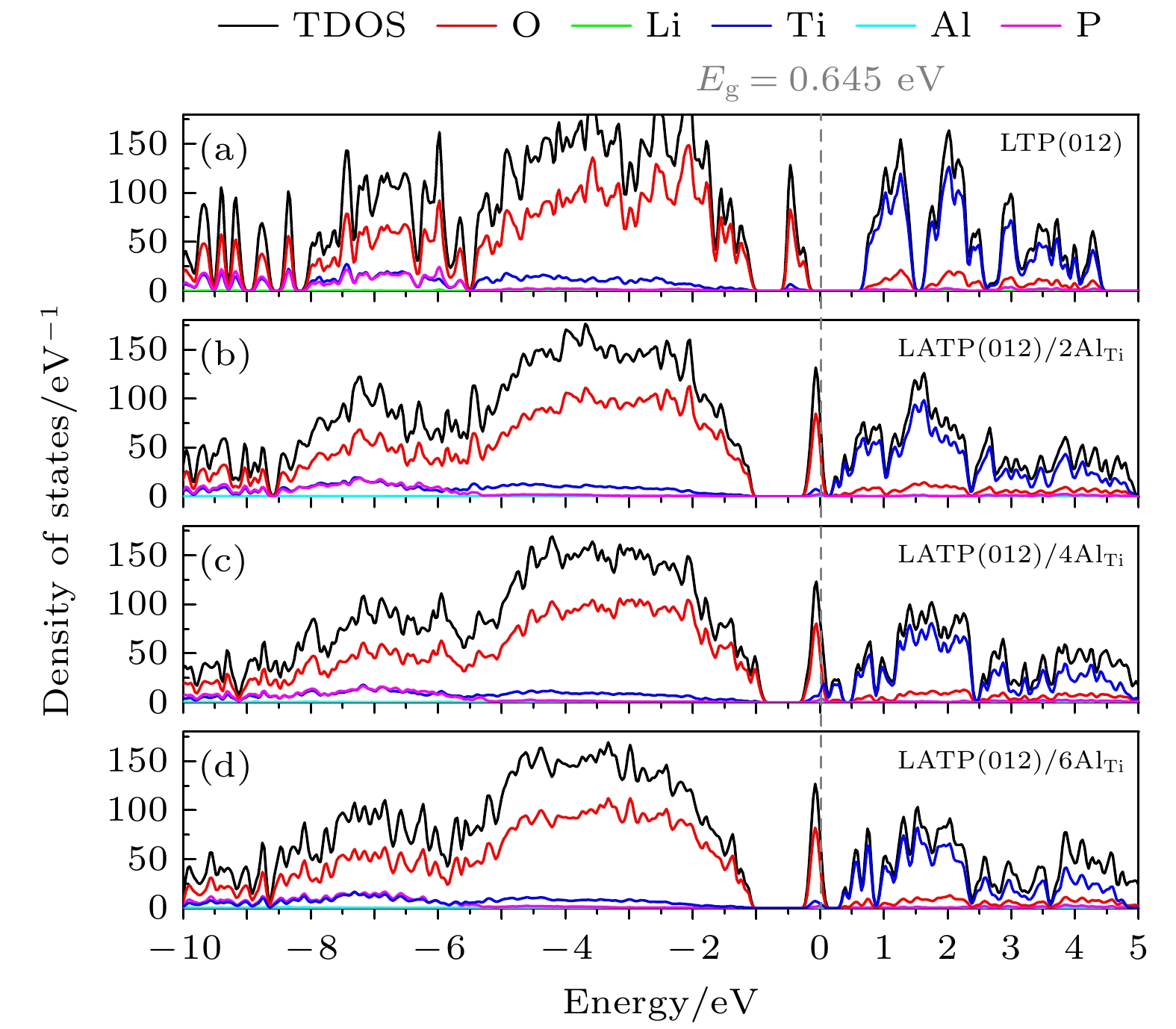
图 4 LTP和LATP表面模型的总态密度(TDOS)和投影态密度(PDOS)
Fig. 4. TDOS and PDOS of LTP and LATP surfaces.
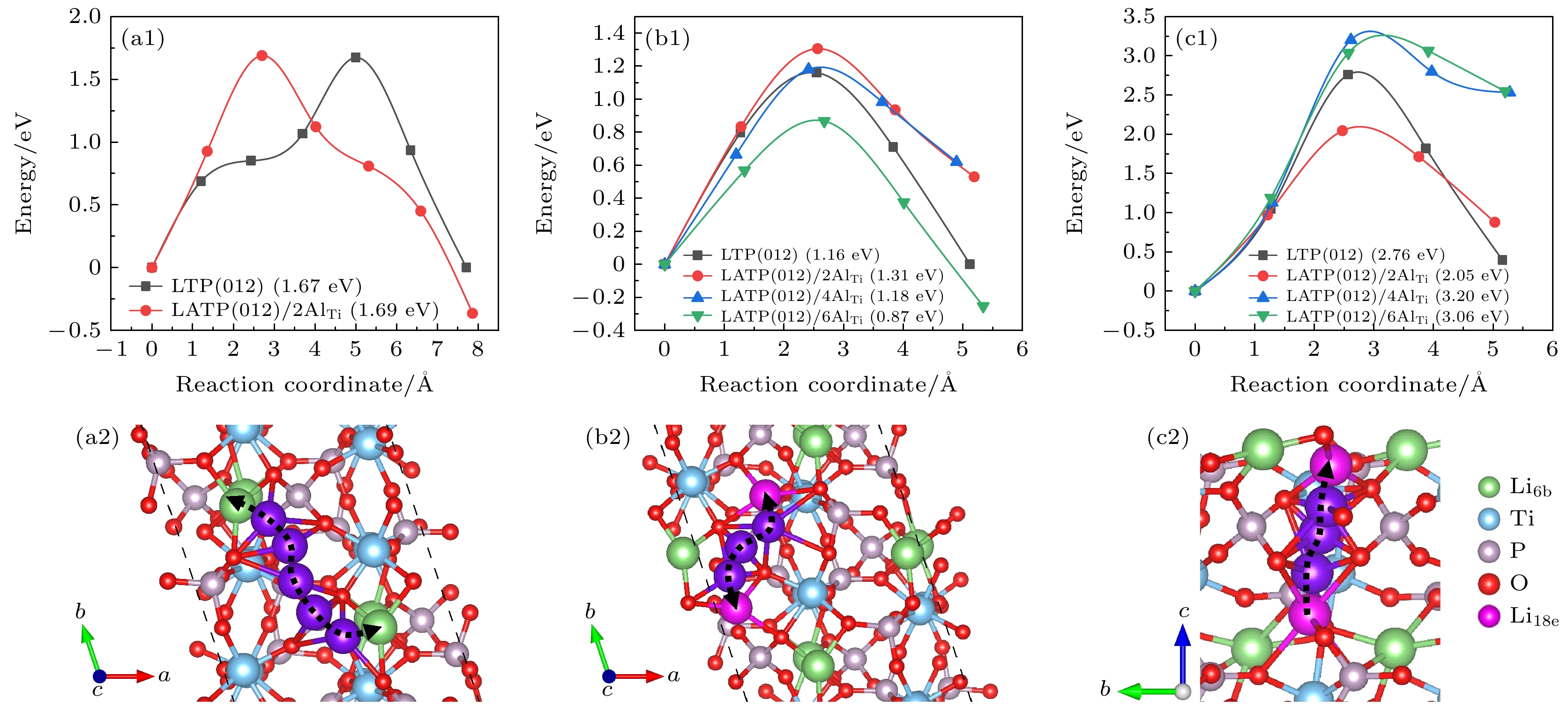
图 5 Li+在LTP和LATP表面的迁移势垒和相应迁移路径 (a1), (a2) 最表层空位迁移; (b1), (b2) 最表层间隙位迁移; (c1), (c2) 次表层到最表层间隙位
Fig. 5. Migration barriers and corresponding migration path for Li+ migration on the LTP and LATP surfaces: (a1), (a2) Vacancy and (b1), (b2) interstitial migration on the outermost layer, (c1), (c2) interstitial migration from the subsurface layer to the outermost layer.
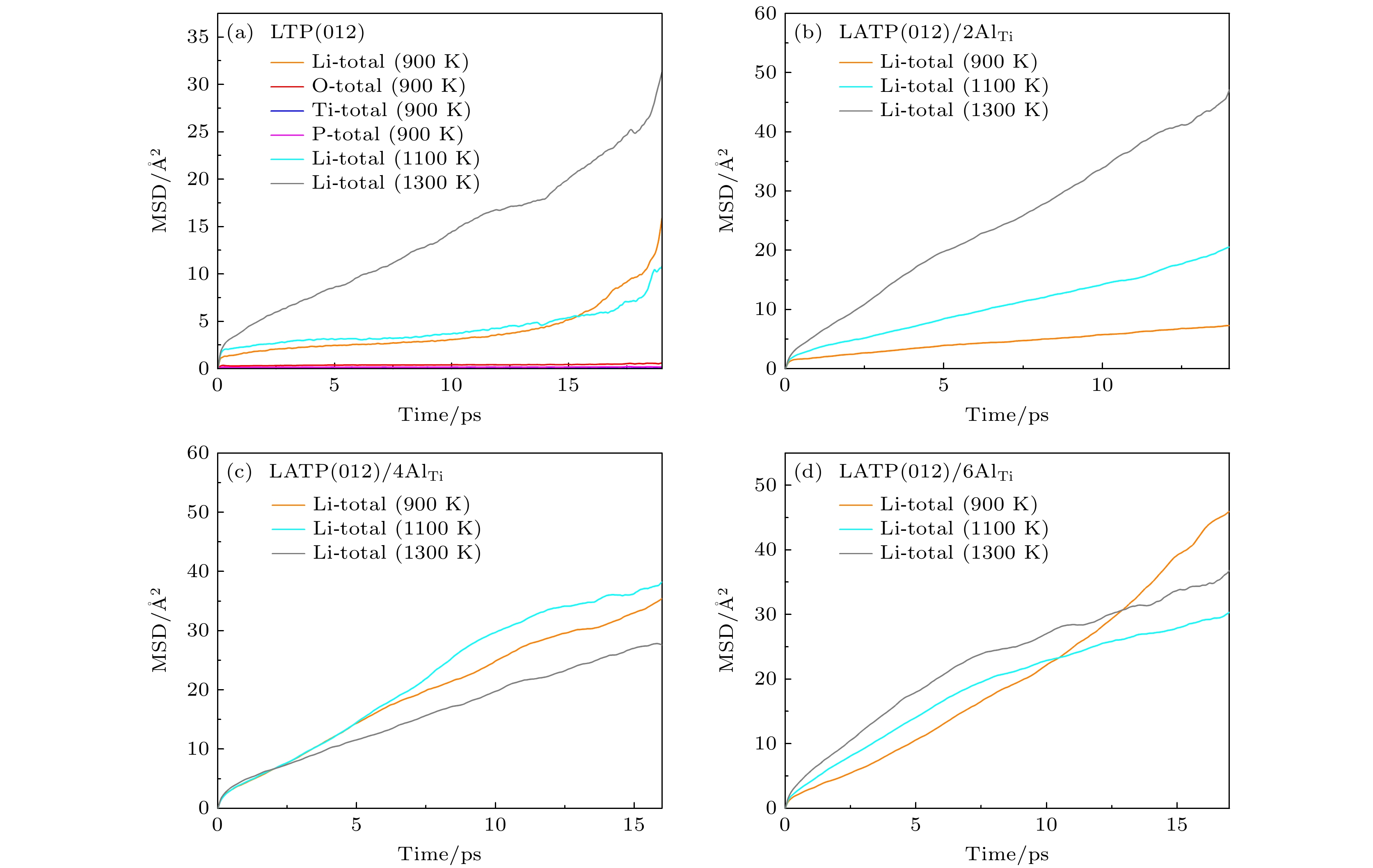
图 6 在温度为900, 1100和1300 K时, Li+在LTP(a)和LATP (012) (b)—(d)表面的MSD随时间的变化曲线图
Fig. 6. The time dependence of MSDs for Li ions at 900, 1100 and 1300 K: (a) LTP(012) surface; (b)–(d) LATP(012) surfaces with different Al content.
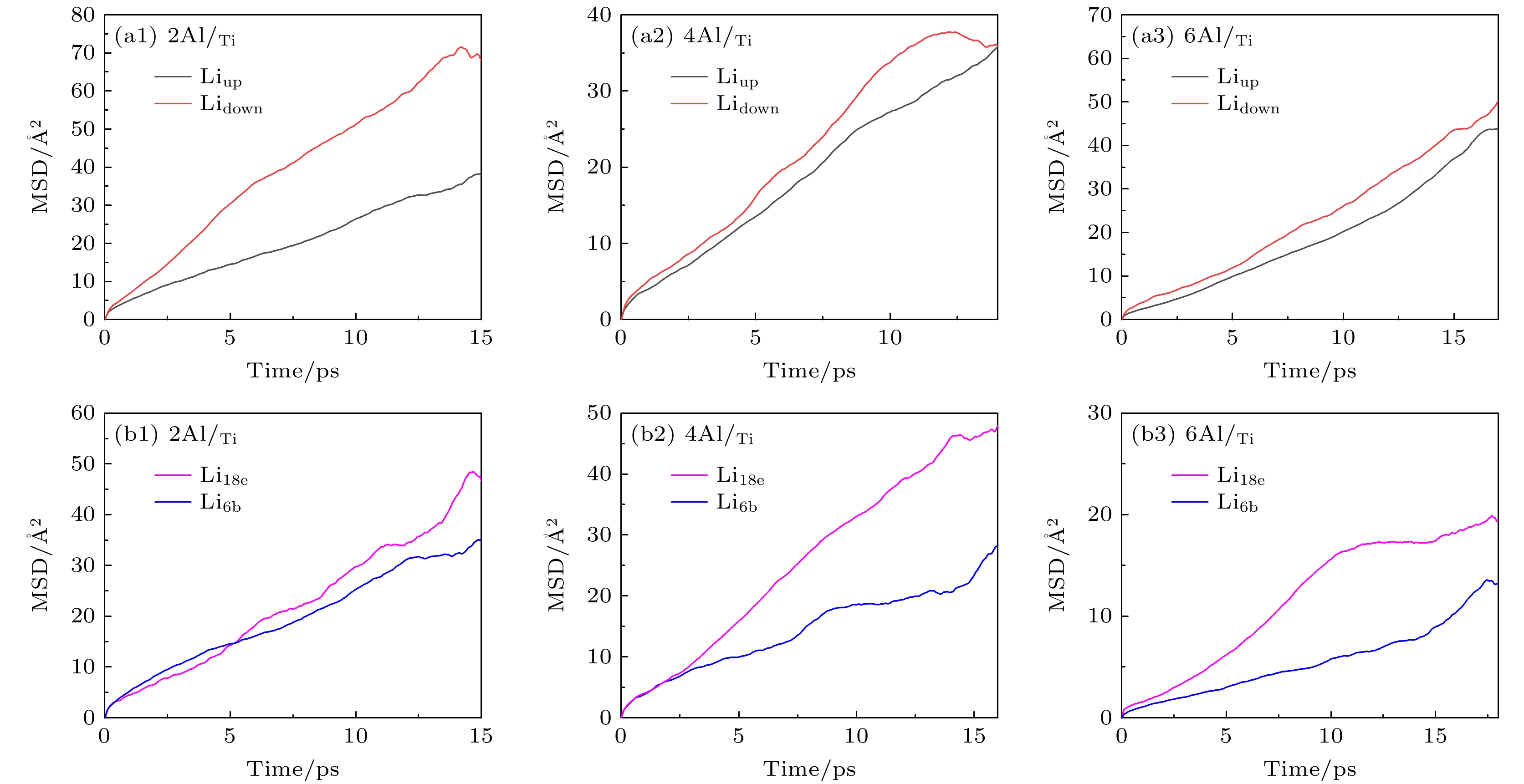
图 7 三种LATP(012)表面结构中不同位置Li的扩散情况 (a1)—(a3)为不同原子层的Li+的MSD图; (b1)—(b3)为最表层中Li6b和Li18e位的Li+的MSD图
Fig. 7. Li diffusion at different positions on three LATP(012) surface: (a1)–(a3) MSD diagrams of Li+ on different atomic layers; (b1)–(b3) MSD diagrams of Li+ at Li6b and Li18e positions on the outermost layer.
表 1 不同晶面指数的LTP表面的表面能 ($ \gamma $)
Table 1. Surface energy ($ \gamma $) of LTP surfaces with different crystal face.
Facets Termination $ \gamma $/(J·m–2) (012) Li— 0.85 (100) O— 1.71 (101) O— 1.70 Ti— 1.88 (001) Li— 2.31 Ti/O— 2.41  下载: 导出CSV
下载: 导出CSV
表 2 不同Al含量LATP(012)表面的表面能 ($ \gamma $)和化学式 (SFs)
Table 2. Surface energy ($ \gamma $) and structural formulas (SFs) of the LATP(012) surface with different Al content.
Surface SFs $ \gamma $/(J·m–2) LATP(012)/2AlTi Li14Al2Ti22P36O144 0.68 LATP(012)/4AlTi Li16Al4Ti20P36O144 0.60 LATP(012)/6AlTi Li18Al6Ti18P36O144 0.43
下载: 导出CSV
表 3 在温度为900, 1100和1300 K时, LTP和LATP(012)表面结构中Li+平均扩散系数和电导率
Table 3. Average Li+ diffusion coefficient and conductivity on the LTP and LATP (012) surfaces at 900, 1100 and 1300 K.
温度/K 结构 扩散系数/
(cm2·S–1)电导率/
(S·cm–1)900 LTP(012) 7.56×10–6 6.80×10–6 LATP(012)/2AlTi 6.59×10–6 7.30×10–6 LATP(012)/4AlTi 3.24×10–5 4.04×10–5 LATP(012)/6AlTi 4.50×10–5 6.21×10–5 1100 LTP(012) 4.81×10–6 3.56×10–6 LATP(012)/2AlTi 2.42×10–5 2.18×10–5 LATP(012)/4AlTi 3.88×10–5 3.95×10–5 LATP(012)/6AlTi 2.56×10–5 2.89×10–5 1300 LTP(012) 2.26×10–5 1.42×10–5 LATP(012)/2AlTi 5.50×10–5 4.21×10–5 LATP(012)/4AlTi 2.36×10–5 2.03×10–5 LATP(012)/6AlTi 3.11×10–5 2.97×10–5
下载: 导出CSV
-
[1] Zhang S, Ma J, Dong S M, Cui G L 2023 Electrochem. Energy Rev. 6 4
Google Scholar
[2] Manthiram A, Yu X W, Wang S F 2017 Nat. Rev. Mater. 2 16103
Google Scholar
[3] Bachman J C, Muy S, Grimaud A, Chang H H, Pour N, Lux S F, Paschos O, Maglia F, Lupart S, Lamp P, Giordano L, Shao-Horn Y 2016 Chem. Rev. 116 140
Google Scholar
[4] Fan L, Wei S Y, Li S Y, Li Q, Lu Y Y 2018 Adv. Energy Mater. 8 1702657
Google Scholar
[5] Zhang Z Z, Shao Y J, Lotsch B, Hu Y S, Li H, Janek J, Nazar L F, Nan C W, Maier J, Armand M, Chen L Q 2018 Energy Environ. Sci. 11 1945
Google Scholar
[6] Zheng F, Kotobuki M, Song S F, Lai M O, Lu L 2018 J. Power Sources 389 198
Google Scholar
[7] Subramanian M, Subramanian R, Clearfield A 1986 Solid State Ion. 18&19 562
Google Scholar
[8] Adachi G y, Imanaka N, Aono H 1996 Adv. Mater. 8 127
Google Scholar
[9] Aono H, Sugimoto E, Sadaoka Y, Imanaka N, Adachi G Y 1990 J. Electrochem. Soc. 137 1023
Google Scholar
[10] Schroeder M, Glatthaar S, Binder J R 2011 Solid State Ion. 201 49
Google Scholar
[11] Mariappan C R, Gellert M, Yada C, Rosciano F, Roling B 2012 Electrochem. Commun. 14 25
Google Scholar
[12] Yin F S, Zhang Z J, Fang Y L, Sun C W 2023 J. Energy Storage 73 12
Google Scholar
[13] Arbi K, Lazarraga M G, Chehimi D B, Ayadi-Trabelsi M, Rojo J M, Sanz J 2004 Chem. Mater. 16 255
Google Scholar
[14] Arbi K, Hoelzel M, Kuhn A, García-Alvarado F, Sanz J 2013 Inorg. Chem. 52 9290
Google Scholar
[15] Monchak M, Hupfer T, Senyshyn A, Boysen H, Chernyshov D, Hansen T, Schell K G, Bucharsky E C, Hoffmann M J, Ehrenberg H 2016 Chem. Mater. 55 2941
Google Scholar
[16] Luo Y Y, Liu X Y, Wen C J, Ning T X, Jiang X X, Lu A X 2023 Appl. Phys. A 129 13
Google Scholar
[17] Liang Y J, Peng C, Kamiike Y, Kuroda K, Okido M 2019 J. Alloy. Compd. 775 1147
Google Scholar
[18] Tian H K, Jalem R, Gao B, Yamamoto Y, Muto S, Sakakura M, Iriyama Y, Tateyama Y 2020 ACS Appl. Mater. Interface 12 54752
Google Scholar
[19] Wu P, Zhou W, Su X, Li J, Su M, Zhou X, Sheldon B W, Lu W 2023 Adv. Energy Mater. 13 2203440
Google Scholar
[20] Stegmaier S, Schierholz R, Povstugar I, Barthel J, Rittmeyer S P, Yu S, Wengert S, Rostami S, Kungl H, Reuter K 2021 Adv. Energy Mater. 11 2100707
Google Scholar
[21] Kresse G, Hafner J 1994 J. Phys.: Condens. Matter 6 8245
Google Scholar
[22] Kresse G, Hafner J 1993 Phys. Rev. B 47 558
Google Scholar
[23] Kresse G, Furthmüller J 1996 Phys. Rev. B 54 11169
Google Scholar
[24] Perdew J P, Burke K, Ernzerhof M 1996 Phys. Rev. Lett. 77 3865
Google Scholar
[25] Perdew J P, Ernzerhof M, Burke K 1996 Chem. Phys. 105 9982
Google Scholar
[26] Pack J D, Monkhorst H J 1977 Phys. Rev. B 16 1748
Google Scholar
[27] Henkelman G, Uberuaga B P, Jónsson H 2000 Chem. Phys. 113 9901
Google Scholar
[28] Nosé S 1984 J. Chem. Phys. 81 511
Google Scholar
[29] Tian H K, Liu Z, Ji Y Z, Chen L Q, Qi Y 2019 Chem. Mater. 31 7351
Google Scholar
[30] 李梅, 钟淑英, 胡军平, 孙宝珍, 徐波 2024 73 362
Google Scholar
Li M, Zhong S Y, Hu J P, Sun B Z, Xu B 2024 Acta Phys. Sin. 73 362
Google Scholar
[31] Han F D, Westover A S, Yue J, Fan X L, Wang F, Chi M F, Leonard D N, Dudney N J, Wang H, Wang C S 2019 Nat. Energy 4 187
Google Scholar
[32] Lang B, Ziebarth B, Elsässer C 2015 Chem. Mater. 27 5040
Google Scholar
[33] Yang K, Chen L K, Ma J B, He Y B, Kang F Y 2021 InfoMat. 3 1195
Google Scholar





 下载:
下载:






计量
- 文章访问数: 3146
- PDF下载量: 61
- 被引次数: 0
