











-
Siligraphene, as a composite of graphene and silicene, has attracted widespread attraction since it has many excellent properties that neither of graphene and silicene possesses. The properties of siligraphene are closely related to the distribution of Si atoms and its structure, but most of the current researches of siligraphene focus on the regular distribution of Si atoms and the planar structure with high symmetry. Therefore, we study in this work all possible Si atoms’ distributions with planar and nonplanar structures for siligraphene g-SiC7 based on density functional theory. At first, 365 kinds of inequivalent Si atoms’ distributions in g-SiC7 are selected out from the 35960 kinds of Si atoms’ distributions, and then for each inequivalent Si atoms’ distribution, a comparison of the stability between the planer and nonplanar structures is made. In terms of the Si distribution, the Si atoms tend to gather together to lower the energy. The more dispersed Si atoms’ distribution usually has appreciably higher energy. In terms of the planarity of the structures, it is found that there are many non-planar structures with significantly lower energy than the planar ones. For all possible Si atoms’ distributions, there are only 8 planar structures which are stable against out-of-plane perturbations. We further study the dynamic, thermodynamic and mechanical stability of the structures with the lowest energies and find that they are stable. The energy band calculation shows that two Dirac valleys still persist in the first Brillouin zone despite their appreciable structure deformation, and a considerable band gap is opened at the Dirac point. We calculate the Berry curvatures and find that the Berry curvatures at the inequivalent valleys are opposite, indicating that the system has valley degree of freedom. Our research shows that siligraphene is more likely to have a buckled structure and a more concentrated silicon atoms’ distribution, and the most stable structures have good electronic properties.
-
Keywords:
- siligraphene /
- silicon distribution /
- planar and nonplanar structures /
- first-principles calculation
[1] Kim S, Ihm J, Choi H J, Son Y W 2008 Phys. Rev. Lett. 100 176802
 Google Scholar
Google Scholar
[2] Lusk M T, Carr L D 2008 Phys. Rev. Lett. 100 175503
Google Scholar
[3] Si N, Zhang F, Jiang W, Zhang Y L 2018 Physica 510 641
Google Scholar
[4] Meng L, Wang Y L, Zhang L Z, Du S X, Wu R T, Li L F, Zhang Y, Li G, Zhou H, Hofer W A 2013 Nano Lett. 13 685
Google Scholar
[5] Li L F, Lu S Z, Pan J B, Qin Z H, Wang Y Q, Wang Y L, Cao G Y, Du S X, Gao H J 2014 Adv. Mater. 26 4820
Google Scholar
[6] Nijamudheen A, Bhattacharjee R, Choudhury S, Datta A 2015 J. Phys. Chem. C 119 3802
Google Scholar
[7] Wang H W, Wu M S, Lei X L, Tian Z F, Xu B, Huang K, Ouyang C Y 2018 Nat. Energy 49 67
[8] Zhou L J, Dong H L, Tretiak S 2020 Nanoscale 12 4269
Google Scholar
[9] Abdullah N R, Mohammed G A, Rashid H O, Gudmundsson V 2020 Phys. Lett. 384 126578
Google Scholar
[10] Liu X B, Shao X F, Yang B, Zhao M W 2018 Nanoscale 10 2108
Google Scholar
[11] Tang X D, Liu W C, Luo C B, Peng X Y, Zhong J X 2019 RSC Adv. 9 12276
Google Scholar
[12] Zhou L J, Zhang Y F, Wu L M 2013 Nano Lett. 13 5431
Google Scholar
[13] Dong H L, Lin B, Gilmore K, Hou T J, Lee S T, Li Y Y 2015 J. Power Sources 299 371
Google Scholar
[14] Zhao M W, Zhang R Q 2014 Phys. Rev. B 89 195427
Google Scholar
[15] Dong H L, Wang L, Zhou L J, Hou T J, Li Y Y 2017 Carbon 113 114
Google Scholar
[16] Dong H L, Zhou L J, Frauenheim T, Hou T J, Lee S T, Li Y Y 2016 Nanoscale 8 6994
Google Scholar
[17] Naqvi S R, Hussain T, Luo W, Ahuja R 2018 Nano Res. 11 3802
Google Scholar
[18] Houmad M, Essaoudi I, Ainane A, El K A, Benyoussef A, Ahuja R 2019 Optik 177 118
Google Scholar
[19] Wu C W, Huang J H, Yao D X 2019 J. Magn. Magn. Mater. 469 306
Google Scholar
[20] Taluja Y, SanthiBhushan B, Yadav S, Srivastava A 2016 Superlattices Microstruct. 98 306
Google Scholar
[21] Sun S, Qi Y, Zhang T Y 2015 Electrochim. Acta 163 296
Google Scholar
[22] Yarmohammadi M, Ebrahimi M R 2019 Phys. Rev. B 100 165409
Google Scholar
[23] Hafner J 2008 J. Comput. Chem. 29 2044
Google Scholar
[24] Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch B M, Walter U 1995 EMBO J. 14 1583
Google Scholar
[25] Rusnak A J, Pinnick E R, Calderon C E, Wang F 2012 J. Chem. Phys. 137 034510
Google Scholar
[26] Choi H C, Park Y C, Kim Y H, Lee Y S 2011 J. Am. Chem. Soc. 133 2084
Google Scholar
[27] Togo A, Tanaka I 2015 Scr. Mater. 108 1
Google Scholar
[28] Ding Y, Wang Y l 2013 J. Phys. Chem. C 117 18266
Google Scholar
[29] Mostofi A A, Yates J R, Pizzi G, Lee Y S, Souza I, Vanderbilt D, Marzar N 2014 Comput. Phys. Commun. 185 2309
Google Scholar
[30] Facio J I, Efremov D, Koepernik K, You J S, Sodemann I, Van D B J 2018 Phys. Rev. Lett. 121 246403
Google Scholar
[31] Shi X Z, He C Y, Pickard C J, Tang C, Zhong J X 2018 Phys. Rev. B 97 014104
Google Scholar
[32] Yin H C, Shi X Z, He C Y, Martinez C M, Li J, Pickard C J, Tang C, Ouyang T, Zhang C X, Zhong J X 2019 Phys. Rev. B 99 041405
Google Scholar
[33] He C Y, Shi X Z, Clark S J, Li J, Pickard C J, Ouyang T, Zhang C X, Tang C, Zhong J X 2018 Phys. Rev. Lett. 121 175701
Google Scholar
[34] Zhou N, Zhou P, Li J, He C Y, Zhong J X 2019 Phys. Rev. B 100 115425
Google Scholar
[35] Li J, Li S, Ouyang T, Zhang C, Tang C, He C, Zhong J 2021 J. Phys. Chem. Lett. 12 732
Google Scholar
[36] Gong Z, Shi X, Li J, Li S, He C, Ouyang T, Zhang C, Tang C, Zhong J 2020 Phys. Rev. B 101 155427
Google Scholar
-
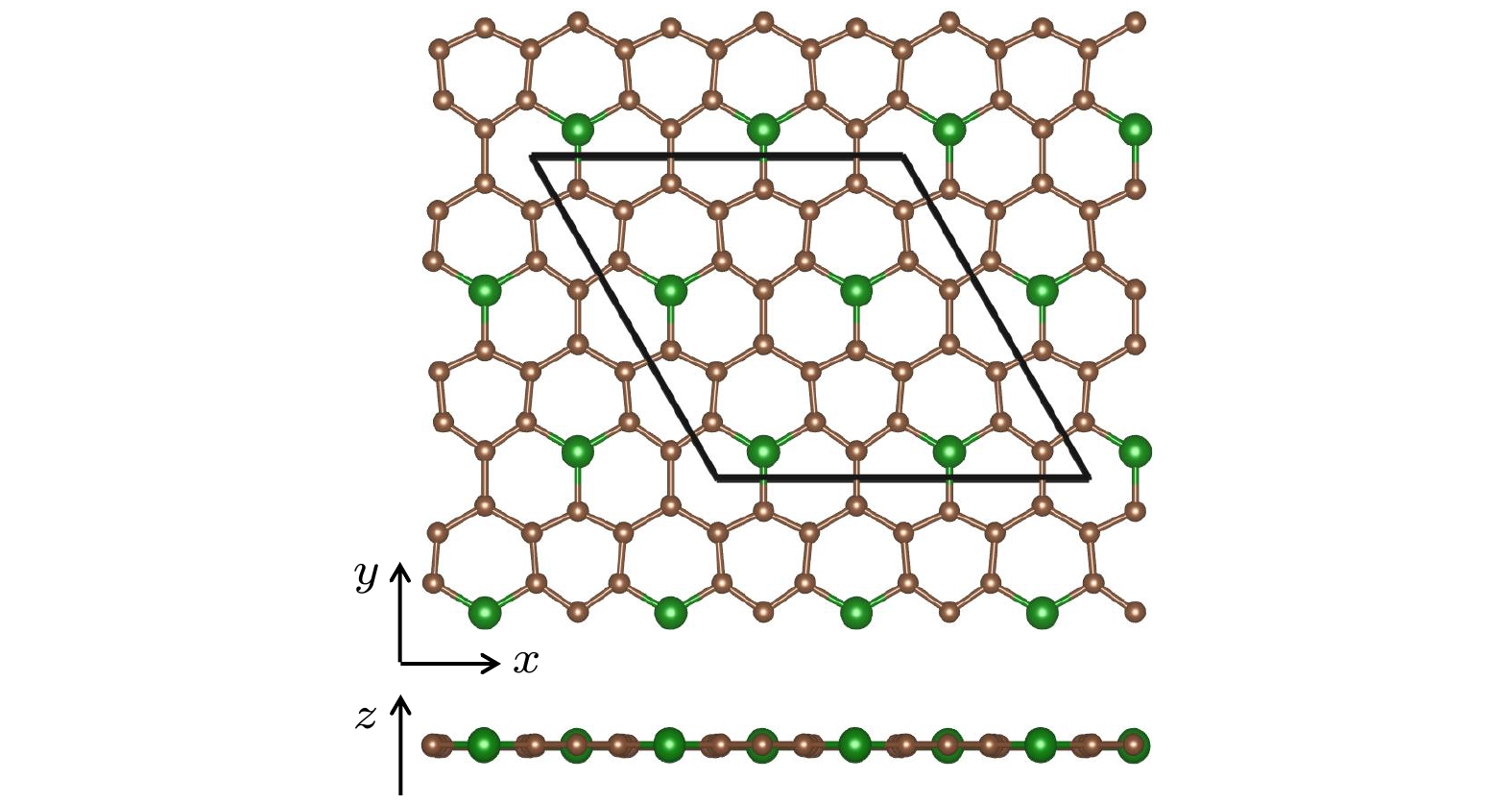

图 2 g-SiC7的总能与Si原子间平均距离的关系, 选取文献[17, 18]中g0-SiC7的总能为参考零点. 图中的罗马数字标记的是能量最低的非平面结构、平面结构和本文中与它们作为比较的一些结构. 插图柱状图代表在在不同能量区间中不等价Si分布在总分布数中的百分比
Figure 2. For the relationship between the total energy of g-SiC7 and the average distance between Si atoms, the total energy of g0-SiC7 in the literature [17, 18] is selected as the reference zero point. The Roman numerals in the figure mark the non-planar structures with the lowest energy, planar structures, and some structures compared with them in this article. The inset histogram represents the percentage of unequal Si distribution in the total distribution in different energy intervals.
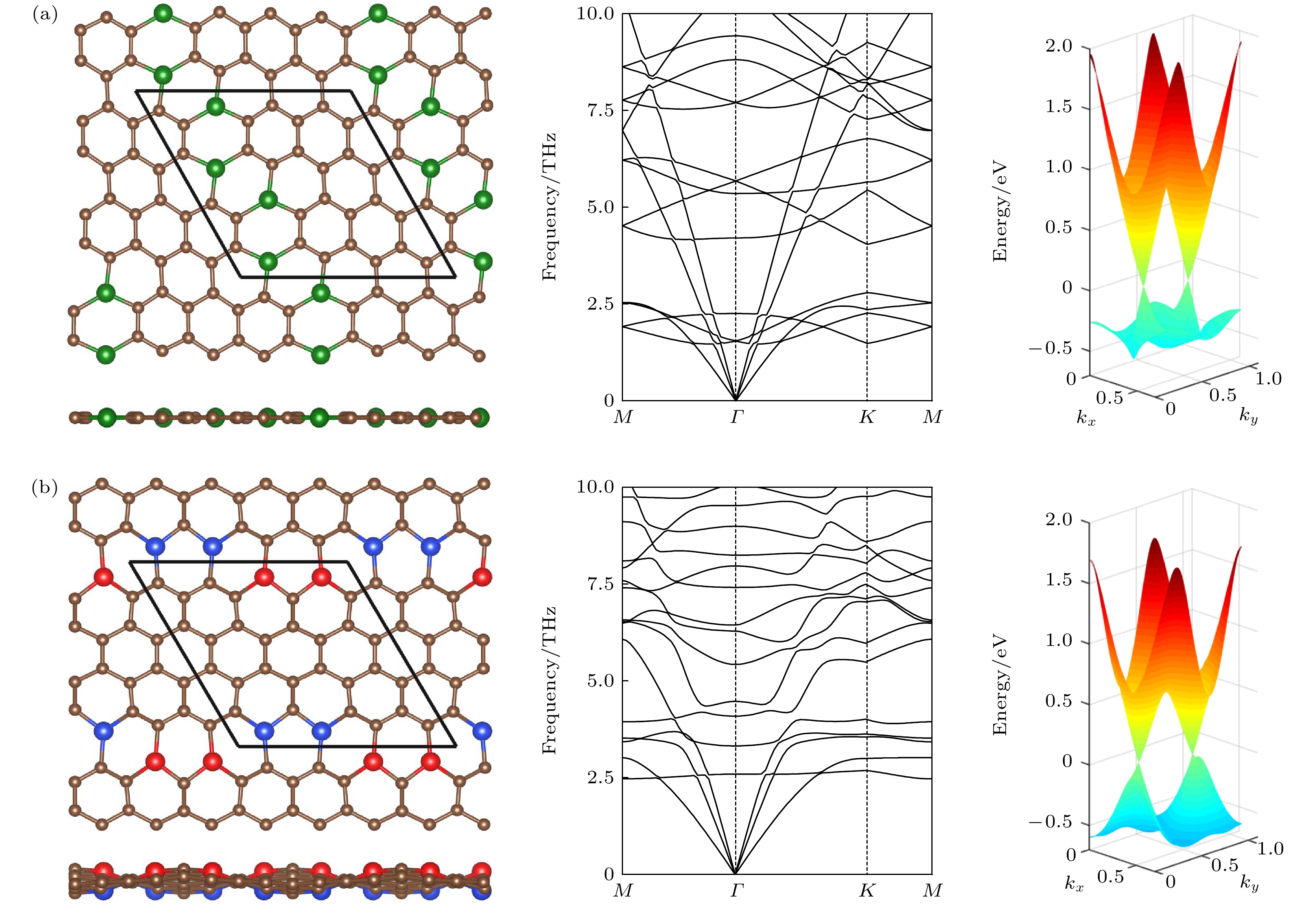
图 3 (a)和(b)中的左图分别对应图2中结构Ⅴ和Ⅳ的俯视图和侧视图, 其中棕色球表示C原子, 红色、绿色和蓝色球分别表示在平面上、平面内和平面下的Si原子, 棱形代表原胞. 中图和右图分别是它们的声子谱和能带
Figure 3. The left panels of (a) and (b) are the top and side views of the structures V and IV in Fig. 2, respectively. The brown spheres stand for C atoms. The red, green and blue spheres denote the Si atoms above, inside and below the plane, respectively. The rhombus denotes the unit cell. The middle and right panels are their phonon spectra and energy bands, respectively.

图 4 (a), (b)和(c)中的左图分别是图2中能量最低结构Ⅰ, Ⅱ和Ⅲ的俯视图和侧视图, 其中棕色球表示C原子, 红色、绿色和蓝色球分别表示在平面上、平面内和平面下的Si原子, 棱形代表原胞, h代表翘曲高度. 中图和右图分别是它们的声子谱和能带. 在能带图中间的颜色梯度图为价带总贝里曲率
Figure 4. The left panels in Fig. (a), (b) and (c) are the top and side views of the structures I, II and III in Fig. 2 with the lowest energies, respectively. The brown spheres stand for C atoms. The red, green and blue spheres denote the Si atoms above, inside and below the plane, respectively. The rhombus denotes the unit cell, h denote buckling height. The middle and right panels are their phonon spectra and energy bands, respectively. The central color gradient plane between the bands shows the calculated total Berry curvature of the valence bands.
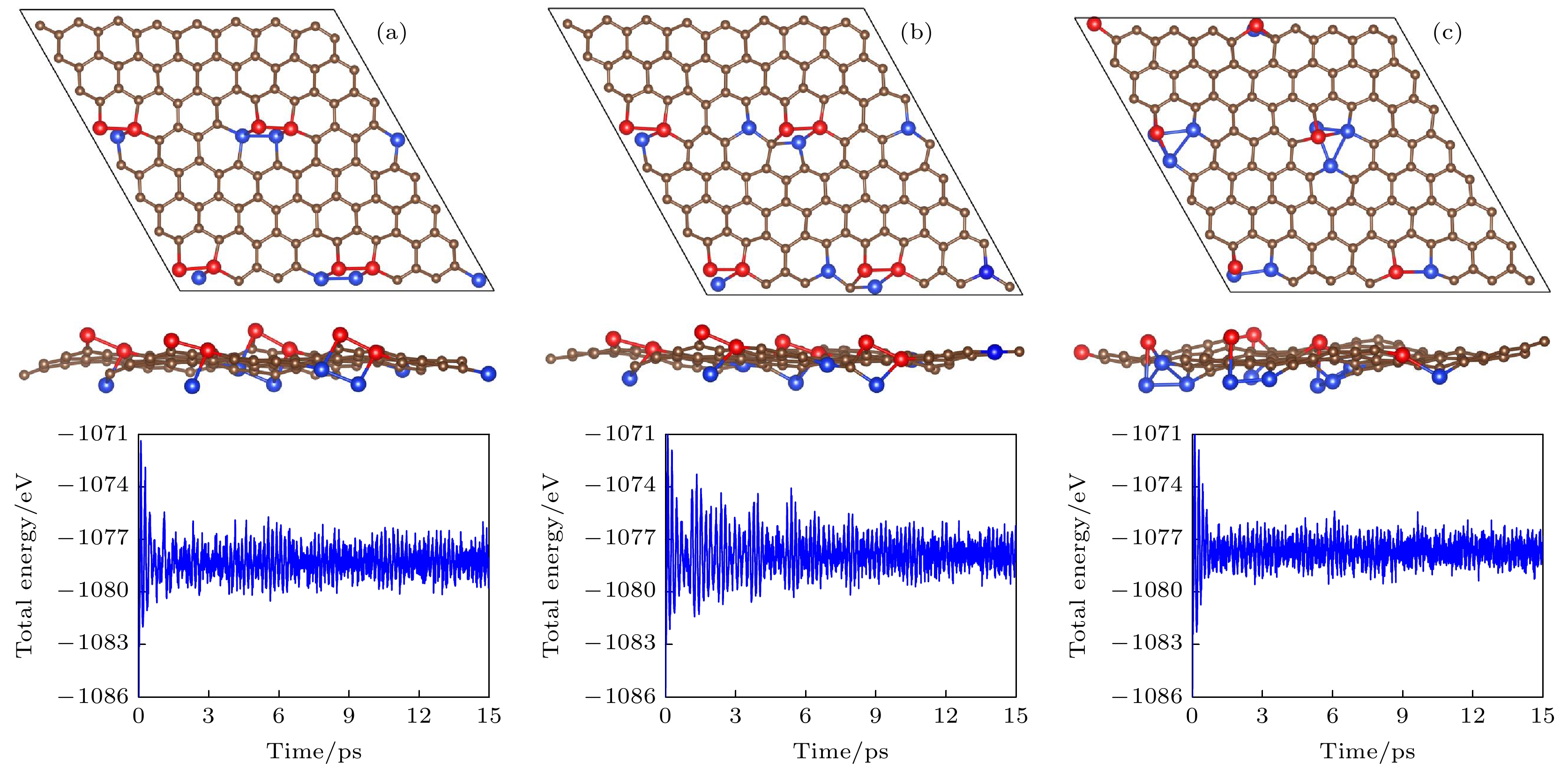
图 5 (a), (b)和(c)分别为图2中g-SiC7的能量最低结构Ⅰ, Ⅱ和Ⅲ在500 K温度下的分子动力学模拟结果. 左图为15 ps后的几何结构, 右图为模拟过程中的能量随时间变化
Figure 5. (a), (b) and (c) are the molecular dynamics simulations (at 500 K) of the three most stable structures I, II and III of g-SiC7 in Fig. 2. The left and right panels show the structures after 15 ps and the total energies vs time, respectively.
表 1 最稳定的三种g-SiC7的总能(以g0-SiC7的总能为参照)、翘曲高度h、带隙和弹性模量, 罗马数字与图2中的对应
Table 1. The total energy (with respect to the total energy of g0-SiC7), buckling height h, band gap and elastic moduli of the three most stable g-SiC7, the Roman numbers correspond to those in Fig. 2.
Structure Ⅰ-SiC7 Ⅱ-SiC7 Ⅲ-SiC7 Energy/(eV per formula unit) –1.85 –1.81 –1.80 h/Å 2.84 2.45 2.63 Band gap/eV 0.02 0.42 0.26 C11/(N·m–1) 237.30 270.45 219.15 C22/(N·m–1) 178.20 209.40 218.55 C12/(N·m–1) 12.30 31.50 21.90 C44/(N·m–1) 93.60 97.65 95.70  DownLoad: CSV
DownLoad: CSV
-
[1] Kim S, Ihm J, Choi H J, Son Y W 2008 Phys. Rev. Lett. 100 176802
Google Scholar
[2] Lusk M T, Carr L D 2008 Phys. Rev. Lett. 100 175503
Google Scholar
[3] Si N, Zhang F, Jiang W, Zhang Y L 2018 Physica 510 641
Google Scholar
[4] Meng L, Wang Y L, Zhang L Z, Du S X, Wu R T, Li L F, Zhang Y, Li G, Zhou H, Hofer W A 2013 Nano Lett. 13 685
Google Scholar
[5] Li L F, Lu S Z, Pan J B, Qin Z H, Wang Y Q, Wang Y L, Cao G Y, Du S X, Gao H J 2014 Adv. Mater. 26 4820
Google Scholar
[6] Nijamudheen A, Bhattacharjee R, Choudhury S, Datta A 2015 J. Phys. Chem. C 119 3802
Google Scholar
[7] Wang H W, Wu M S, Lei X L, Tian Z F, Xu B, Huang K, Ouyang C Y 2018 Nat. Energy 49 67
[8] Zhou L J, Dong H L, Tretiak S 2020 Nanoscale 12 4269
Google Scholar
[9] Abdullah N R, Mohammed G A, Rashid H O, Gudmundsson V 2020 Phys. Lett. 384 126578
Google Scholar
[10] Liu X B, Shao X F, Yang B, Zhao M W 2018 Nanoscale 10 2108
Google Scholar
[11] Tang X D, Liu W C, Luo C B, Peng X Y, Zhong J X 2019 RSC Adv. 9 12276
Google Scholar
[12] Zhou L J, Zhang Y F, Wu L M 2013 Nano Lett. 13 5431
Google Scholar
[13] Dong H L, Lin B, Gilmore K, Hou T J, Lee S T, Li Y Y 2015 J. Power Sources 299 371
Google Scholar
[14] Zhao M W, Zhang R Q 2014 Phys. Rev. B 89 195427
Google Scholar
[15] Dong H L, Wang L, Zhou L J, Hou T J, Li Y Y 2017 Carbon 113 114
Google Scholar
[16] Dong H L, Zhou L J, Frauenheim T, Hou T J, Lee S T, Li Y Y 2016 Nanoscale 8 6994
Google Scholar
[17] Naqvi S R, Hussain T, Luo W, Ahuja R 2018 Nano Res. 11 3802
Google Scholar
[18] Houmad M, Essaoudi I, Ainane A, El K A, Benyoussef A, Ahuja R 2019 Optik 177 118
Google Scholar
[19] Wu C W, Huang J H, Yao D X 2019 J. Magn. Magn. Mater. 469 306
Google Scholar
[20] Taluja Y, SanthiBhushan B, Yadav S, Srivastava A 2016 Superlattices Microstruct. 98 306
Google Scholar
[21] Sun S, Qi Y, Zhang T Y 2015 Electrochim. Acta 163 296
Google Scholar
[22] Yarmohammadi M, Ebrahimi M R 2019 Phys. Rev. B 100 165409
Google Scholar
[23] Hafner J 2008 J. Comput. Chem. 29 2044
Google Scholar
[24] Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch B M, Walter U 1995 EMBO J. 14 1583
Google Scholar
[25] Rusnak A J, Pinnick E R, Calderon C E, Wang F 2012 J. Chem. Phys. 137 034510
Google Scholar
[26] Choi H C, Park Y C, Kim Y H, Lee Y S 2011 J. Am. Chem. Soc. 133 2084
Google Scholar
[27] Togo A, Tanaka I 2015 Scr. Mater. 108 1
Google Scholar
[28] Ding Y, Wang Y l 2013 J. Phys. Chem. C 117 18266
Google Scholar
[29] Mostofi A A, Yates J R, Pizzi G, Lee Y S, Souza I, Vanderbilt D, Marzar N 2014 Comput. Phys. Commun. 185 2309
Google Scholar
[30] Facio J I, Efremov D, Koepernik K, You J S, Sodemann I, Van D B J 2018 Phys. Rev. Lett. 121 246403
Google Scholar
[31] Shi X Z, He C Y, Pickard C J, Tang C, Zhong J X 2018 Phys. Rev. B 97 014104
Google Scholar
[32] Yin H C, Shi X Z, He C Y, Martinez C M, Li J, Pickard C J, Tang C, Ouyang T, Zhang C X, Zhong J X 2019 Phys. Rev. B 99 041405
Google Scholar
[33] He C Y, Shi X Z, Clark S J, Li J, Pickard C J, Ouyang T, Zhang C X, Tang C, Zhong J X 2018 Phys. Rev. Lett. 121 175701
Google Scholar
[34] Zhou N, Zhou P, Li J, He C Y, Zhong J X 2019 Phys. Rev. B 100 115425
Google Scholar
[35] Li J, Li S, Ouyang T, Zhang C, Tang C, He C, Zhong J 2021 J. Phys. Chem. Lett. 12 732
Google Scholar
[36] Gong Z, Shi X, Li J, Li S, He C, Ouyang T, Zhang C, Tang C, Zhong J 2020 Phys. Rev. B 101 155427
Google Scholar


 DownLoad:
DownLoad:




Catalog
Metrics
- Abstract views: 6574
- PDF Downloads: 113
- Cited By: 0
