










-
Au/CeO2(111)作为一种重要的催化剂体系, 在催化氧化、水气转换反应等多个领域展现出优异的催化性能. 为了深入揭示其催化机理, 特别是在原子尺度上理解活性组分的相互作用. 本文采用密度泛函理论(DFT+U)计算方法, 构建了Au/CeO2(111)体系的原子尺度模型, 通过计算该模型的吸附能、差分电荷密度、巴德电荷以及态密度, 揭示了Au/CeO2(111)的表面吸附行为. 在CeO2(111)的平面区域内, 经过结构弛豫与优化, 确定了5个Au吸附位点. 其中最为稳定的吸附位点并非传统上认为的氧顶位, 而是氧-氧桥位. 在这种吸附构型下, 电荷从Au向Ce4+转移, 导致Ce4+被还原为Ce3+, 伴随着显著的电荷转移现象. 过去的研究更多地关注了平面区域的吸附行为, 而忽视了台阶边缘区域在催化过程中的重要性. 因此, 本研究进一步扩展了研究范围, 深入探讨了4种不同台阶结构对Au吸附的影响, 其中, Type II*和Type III台阶因高度欠配位的Ce原子增强了对Au原子的吸附, 特别是Type III台阶通过显著的电荷转移成为Au的首选吸附位点. 本研究通过构建更全面的Au/CeO2模型, 突破了以往仅关注平面吸附的局限性, 揭示了Au/CeO2在台阶边缘的吸附机制, 为深入理解Au/CeO2(111)的催化机理提供了新的视角.
-
关键词:
- Au/CeO2(111) /
- 电荷转移 /
- 吸附能 /
- 第一性原理计算
Au/CeO2(111), as an important catalyst system, has demonstrated excellent catalytic performances in a variety of fields such as the catalytic oxidation and the water-gas shift reactions. In order to reveal in depth the Au/CeO2(111) catalytic mechanism, especially to understand the interaction of the active components on an atomic scale, in this work, the adsorption properties on the Au/CeO2(111) surface are investigated by calculating the adsorption energy, differential charge density, Bader charge, and the density of states by using density functional theory (DFT+U). First, five adsorption sites of Au/CeO2(111) are identified in the planar region of CeO2(111), and the most stable adsorption configuration is found to be located at the bridging position between surface oxygen atoms (the oxygen-oxygen bridging site), which suggests that Au interacts more closely with the oxygen-oxygen bridging sites. Further, the differential charge density and Bader charge reveal the charge transfer mechanism in the adsorption process. Specifically, the Au atoms are oxidized into Au+, while the Ce4+ ions in the second nearest neighbor of Au are reduced to Ce3+, and the adsorption process is accompanied by a charge transfer phenomenon. Au also exhibits a unique adsorption behavior in the CeO2(111) step-edge region, where a highly under-allocated environment is formed due to the decrease in the coordination number of atoms in the step edge, which enhances the adsorption of Au in a highly under-allocated environment. The adsorption of Au at the step edge is enhanced by the lower coordinated environment due to the reduced coordination number of the atoms at the step edge. By comparing four different types of step structures (Type I, Type II, Type II*, and Type III), it is found that the higher adsorption energy of Au at Type II* site and that at Type III site are both mainly due to the lower coordinated state of Ce atoms at these sites. Charge transfer is also particularly pronounced at the Type III sites. It is also accompanied by electron transferring from Au to Ce4+ ions, making Type III the preferred adsorption site for Au atoms. By constructing a more comprehensive Au/CeO2 model, this study breaks through the previous limitation of focusing only on planar adsorption and reveals the adsorption mechanism of Au/CeO2 at the edge of the step, which provides a new perspective for understanding in depth the catalytic mechanism of Au/CeO2(111).-
Keywords:
- Au/CeO2(111) /
- charge transfer /
- adsorption energy /
- first principles calculations
[1] Haruta M, Kobayashi T, Sano H, Yamada N 1987 Chem. Lett. 16 405
 Google Scholar
Google Scholar
[2] Li Y J, Wen H, Zhang Q, Adachi Y, Arima E, Kinoshita Y, Nomura H, Ma Z M, Kou L, Tsukuda Y, Naitoh Y, Sugawara Y, Xu R, Cheng Z 2018 Ultramicroscopy 191 51
Google Scholar
[3] Zielinski M, Juszczyk W, Kaszkur Z 2022 RSC Adv. 12 5312
Google Scholar
[4] Berrichi A, Bailiche Z, Bachir R 2022 Res. Chem. Intermed. 48 4119
Google Scholar
[5] Megías-Sayago C, Lolli A, Ivanova S, Albonetti S, Cavani F, Odriozola J A 2019 Catal. Today 333 169
Google Scholar
[6] Li Q L, Xie W, Chen G Q, Li Y F, Huang Y J, Chen X D 2015 Nano Res. 8 3075
Google Scholar
[7] Liu H P, Cao Z L, Yang S Y, Ren Q Y, Dong Z J, Liu W, Li Z A, Chen X, Luo L L 2024 Nano Res. 17 4986
Google Scholar
[8] Kim C, Thompson L 2006 J. Catal. 244 248
Google Scholar
[9] Chen Y, Hu P, Lee M H, Wang H 2008 Surf. Sci. 602 1736
Google Scholar
[10] Hernández N C, Grau-Crespo R, De Leeuw N H, Sanz J Fdez 2009 Phys. Chem. Chem. Phys. 11 5246
Google Scholar
[11] Zhang C, Michaelides A, King D A, Jenkins S J 2010 J. Am. Chem. Soc. 132 2175
Google Scholar
[12] Engel J, Francis S, Roldan A 2019 Phys. Chem. Chem. Phys. 21 19011
Google Scholar
[13] Shu P L, Tian X, Guo Q, Ren X, Zhao B, Wen H F, Tang J, Li Y J, Yasuhiro S, Ma Z M, Liu J 2024 Phys. Scr. 99 105990
Google Scholar
[14] Teng B T, Wu F M, Huang W X, Wen X D, Zhao L H, Luo M F 2012 ChemPhysChem 13 1261
Google Scholar
[15] Lu Y, Quardokus R, Lent C S, Justaud F, Lapinte C, Kandel S A 2010 J. Am. Chem. Soc. 132 13519
Google Scholar
[16] Lustemberg P G, Pan Y, Shaw B J, Grinter D, Pang C, Thornton G, Pérez R, Ganduglia-Pirovano M V, Nilius N 2016 Phys. ReV. Lett. 116 236101
Google Scholar
[17] Fu Q, Saltsburg H, Flytzani-Stephanopoulos M 2003 Science 301 935
Google Scholar
[18] Bezkrovnyi O, Bruix A, Blaumeiser D, Piliai L, Schötz S, Bauer T, Khalakhan I, Skála T, Matvija P, Kraszkiewicz P, Pawlyta M, Vorokhta M, Matolínová I, Libuda J, Neyman K M, Kȩpiński L 2022 Chem. Mater. 34 7916
Google Scholar
[19] Tibiletti D, Amieiro-Fonseca A, Burch R, Chen Y, Fisher J M, Goguet A, Hardacre C, Hu P, Thompsett D 2005 J. Phys. Chem. B 109 22553
Google Scholar
[20] 冯婕, 郭强, 舒鹏丽, 温阳, 温焕飞, 马宗敏, 李艳君, 刘俊, 伊戈尔·弗拉基米罗维奇·雅明斯基 2023 72 110701
Google Scholar
Feng J, Guo Q, Shu P L, Wen Y, Wen H F, Ma Z M, Li Y J, Liu J 2023 Acta Phys. Sin. 72 110701
Google Scholar
[21] Perdew J P, Burke K, Ernzerhof M 1996 Phys. ReV. Lett. 77 3865
Google Scholar
[22] Cococcioni M, De Gironcoli S 2005 Phys. Rev. B 71 035105
Google Scholar
[23] Zheng Z Y, Wang D, Zhang Y, Yang F, Gong X Q 2020 Chin. J. Cata. 41 1360
Google Scholar
[24] Wilson E L, Grau-Crespo R, Pang C L, Cabailh G, Chen Q, Purton J A, Catlow C R A, Brown W A, De Leeuw N H, Thornton G 2008 J. Phys. Chem. C 112 10918
Google Scholar
[25] Ma Z M, Shi Y B, Mu J L, Qu Z, Zhang X M, Li Q, Liu J 2017 Appl. Surf. Sci. 394 472
Google Scholar
[26] Owen C J, Jenkins S J 2021 J. Chem. Phys. 154 164703
Google Scholar
[27] Chen L J, Tang Y, Cui L, Ouyang C, Shi S 2013 J. of Power Sources 234 69
Google Scholar
[28] Kim H Y, Henkelman G 2013 J. Phys. Chem. Lett. 4 216
Google Scholar
[29] Kozlov S M, Neyman K M 2014 Phys. Chem. Chem. Phys. 16 7823
Google Scholar
[30] Olbrich R, Pieper H H, Oelke R, Wilkens H, Wollschläger J, Zoellner M H, Schroeder T, Reichling M 2014 Appl. Phys. Lett. 104 081910
Google Scholar
[31] Barth C, Laffon C, Olbrich R, Ranguis A, Parent Ph, Reichling M 2016 Sci Rep. 6 21165
Google Scholar
[32] Shu P L, Guo Q, Tian X, Wei J, Qu Z, Ren X, Wen H F, Tang J, Li Y J, Sugawara Y, Ma Z M, Liu J 2024 Surf. Interfaces 51 104738
Google Scholar
[33] Kozlov S M, Viñes F, Nilius N, Shaikhutdinov S, Neyman K M 2012 J. Phys. Chem. Lett. 3 1956
Google Scholar
[34] Chu D R, Wang Z Q, Gong X Q 2022 Surf. Sci 722 122096
Google Scholar
[35] 温焕飞, 菅原康弘, 李艳君 2020 69 210701
Google Scholar
Wen H F, Sugawara Y, Li Y J 2020 Acta Phys. Sin. 69 210701
Google Scholar
[36] Zhou H, Wang D, Gong X Q 2020 Phys. Chem. Chem. Phys. 22 7738
Google Scholar
[37] Zhou C Y, Wang D, Gong X Q 2019 Phys. Chem. Chem. Phys. 21 19987
Google Scholar
[38] Piliai L, Matvija P, Dinhová T N, Khalakhan I, Skála T, Doležal Z, Bezkrovnyi O, Kepinski L, Vorokhta M, Matolínová I 2022 ACS Appl. Mater. Interfaces 14 56280
Google Scholar
[39] Janssen E M W, Wiegers G A 1978 J. Less-Common Met. 57 47
Google Scholar
-
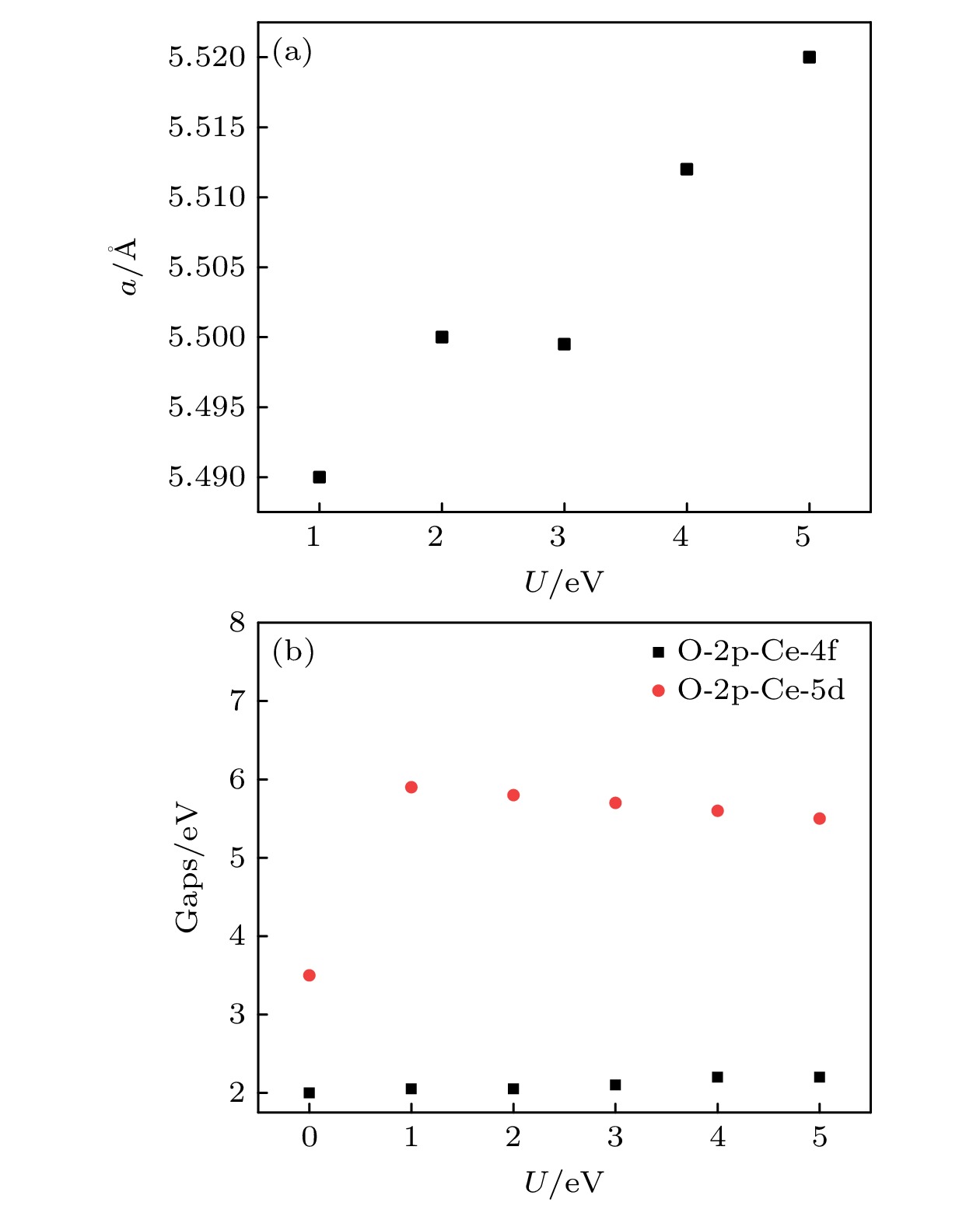
图 1 (a) CeO2晶格常数随U值的变化; (b) CeO2的带隙随U值的变化
Fig. 1. (a) The change of lattice constant of CeO2 with U; (b) the change of gaps of CeO2 with U.
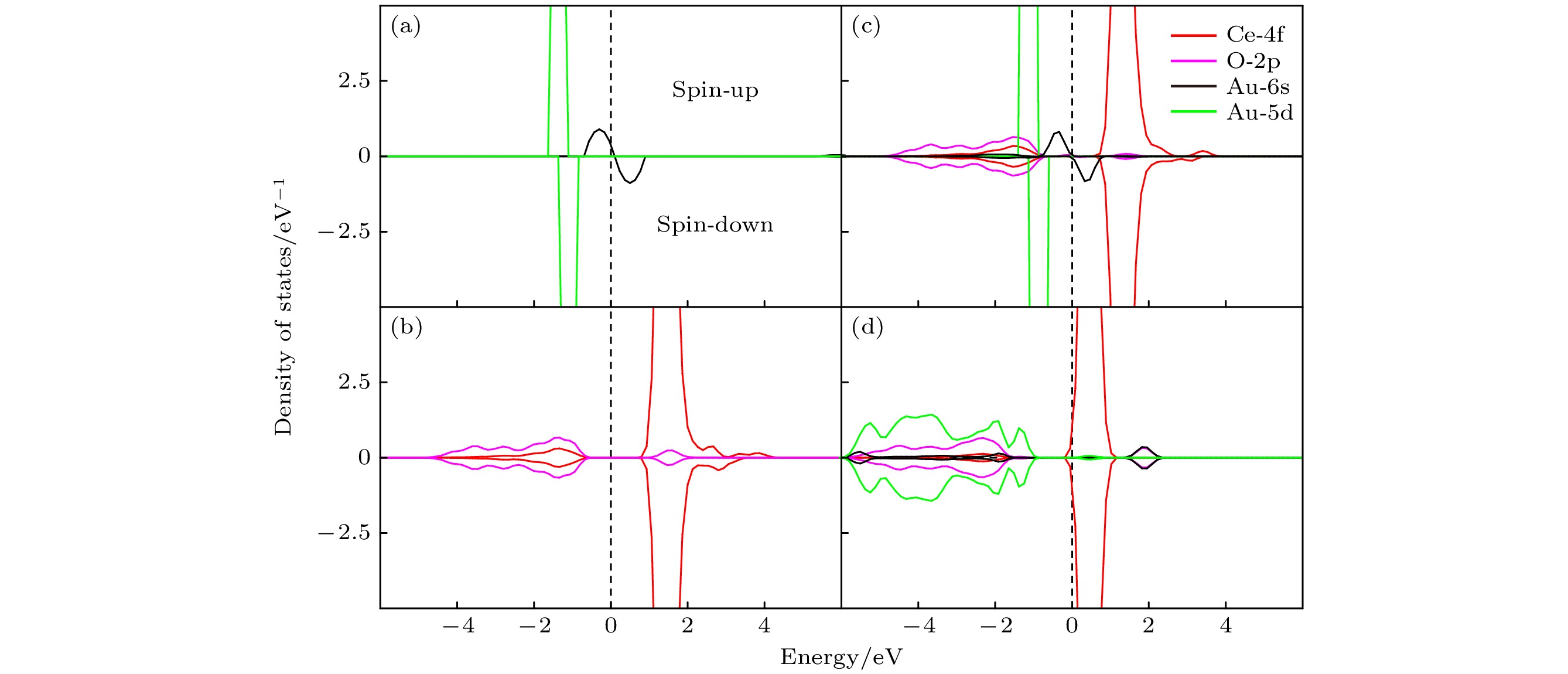
图 4 (a)纯金(Au)的态密度; (b)纯净的CeO2(111) 表面的态密度; (c) Au/CeO2(111) 表面铈顶位的态密度; (d) Au/CeO2(111) 表面氧-氧桥位的态密度, spin-up 和 spin-down 分别代表自旋向上和自旋向下的电子态
Fig. 4. Presents the DOS for various systems: (a) pure gold (Au); (b) pristine CeO2(111) surface; (c) Au/CeO2(111) surface at the cerium top site; (d) Au/CeO2(111) surface at the oxygen-oxygen bridge site, within each plot, spin-up and spin-down represent the electronic states with spin aligned upwards and downwards.
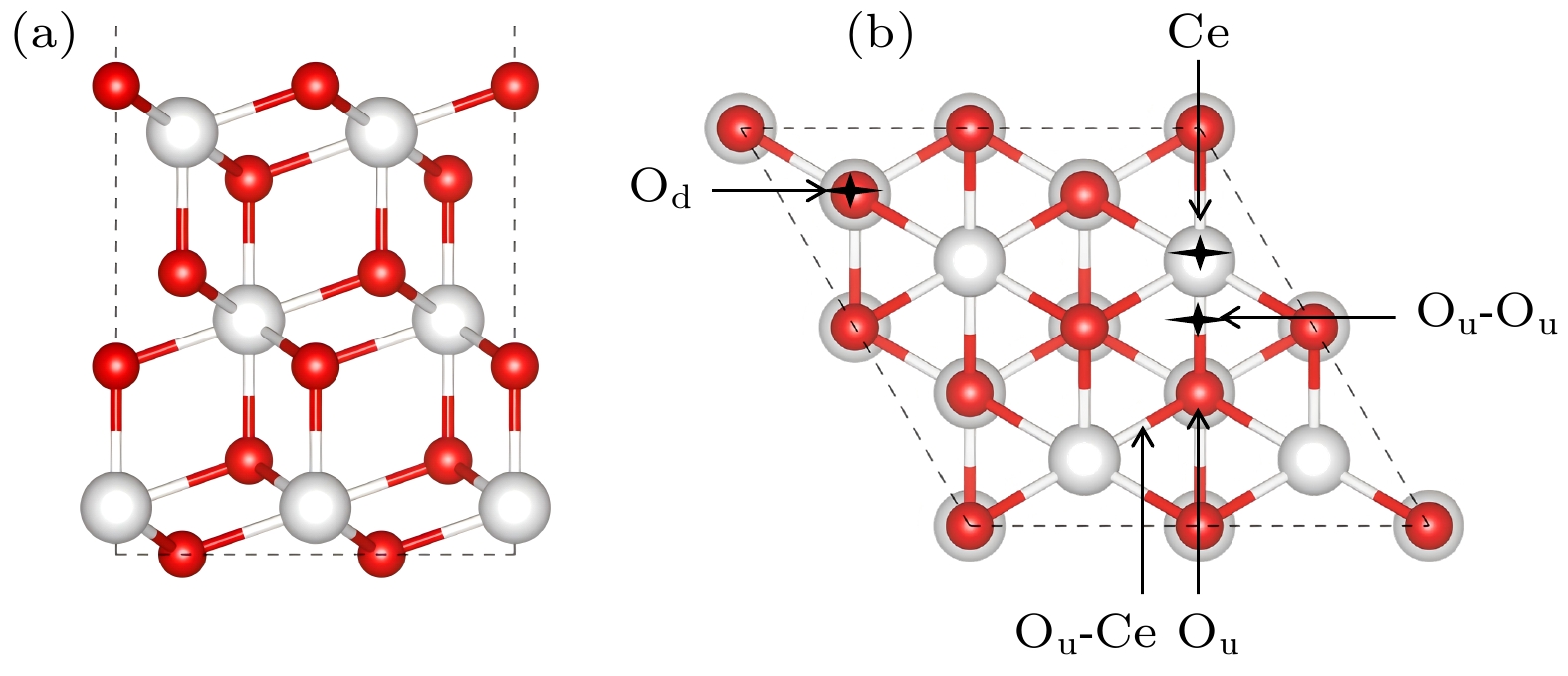
图 2 CeO2(111)表面p(2×2)的结构, 以及Au原子的潜在吸附位点 (a)斜视图; (b)俯视图, 红色和白色球体分别代表O和Ce
Fig. 2. The structure of p(2×2) on CeO2(111) surface and the potential adsorption sites of Au atoms: (a) Side view; (b) top view, the red and white spheres represent O and Ce.
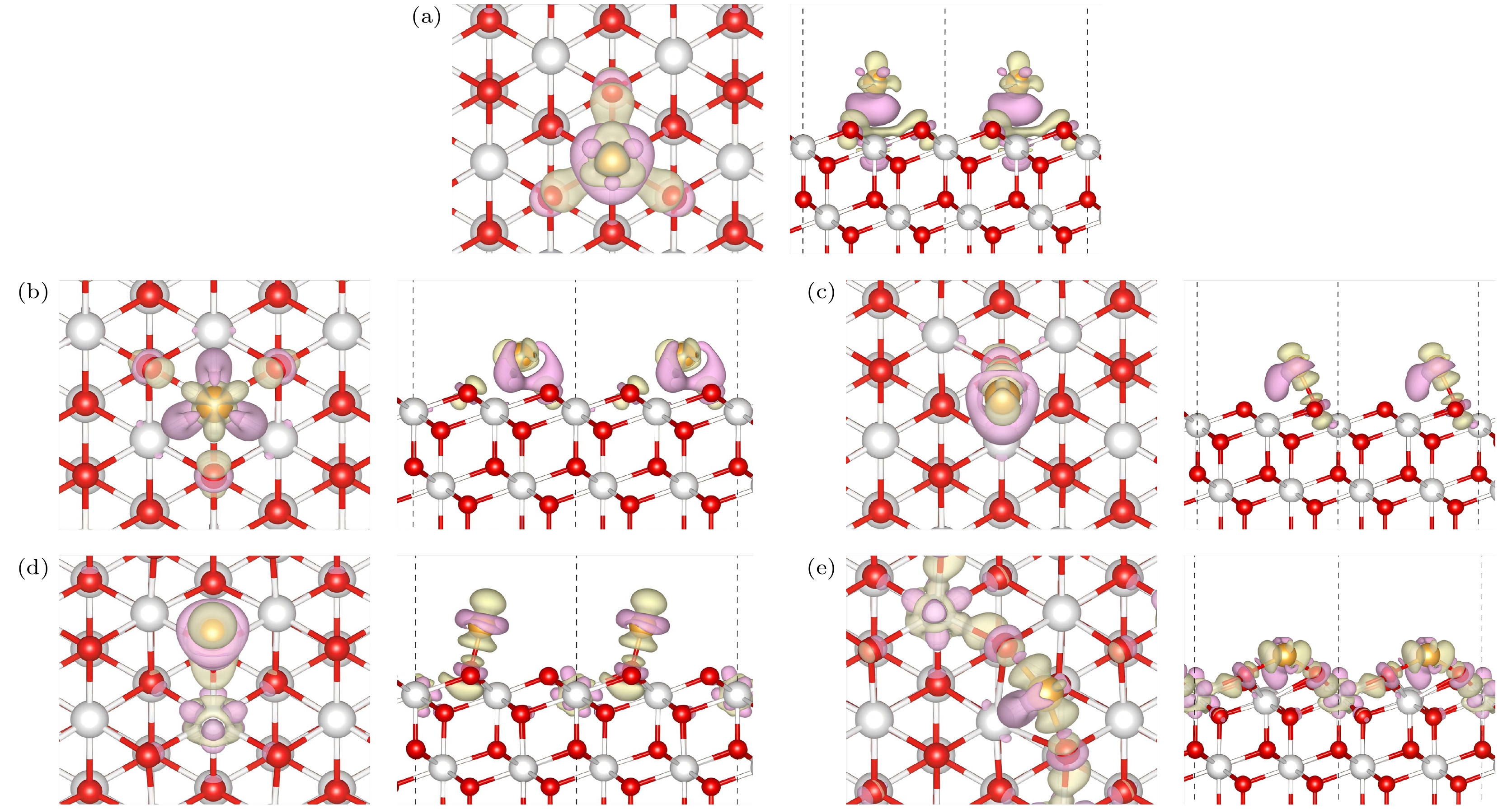
图 3 Au/CeO2(111)表面上的差分电荷密度 (a)铈顶位(Ce); (b)次氧位(Od); (c)顶氧-铈位(Ou-Ce); (d)氧顶位(Ou); (e)氧-氧桥位(Ou-Ou), 图中紫色部分表示该位置电荷减少, 绿色部分表示电荷的增加
Fig. 3. Differential charge density on the Au/CeO2(111) surface: (a) Cerium top site (Ce); (b) sub-oxygen site (Od); (c) top oxygen-cerium site (Ou-Ce); (d) oxygen top site (Ou); (e) oxygen-oxygen bridge site (Ou-Ou), the purple regions in the figure indicate a decrease in charge at that location, while the green regions indicate an increase in charge.
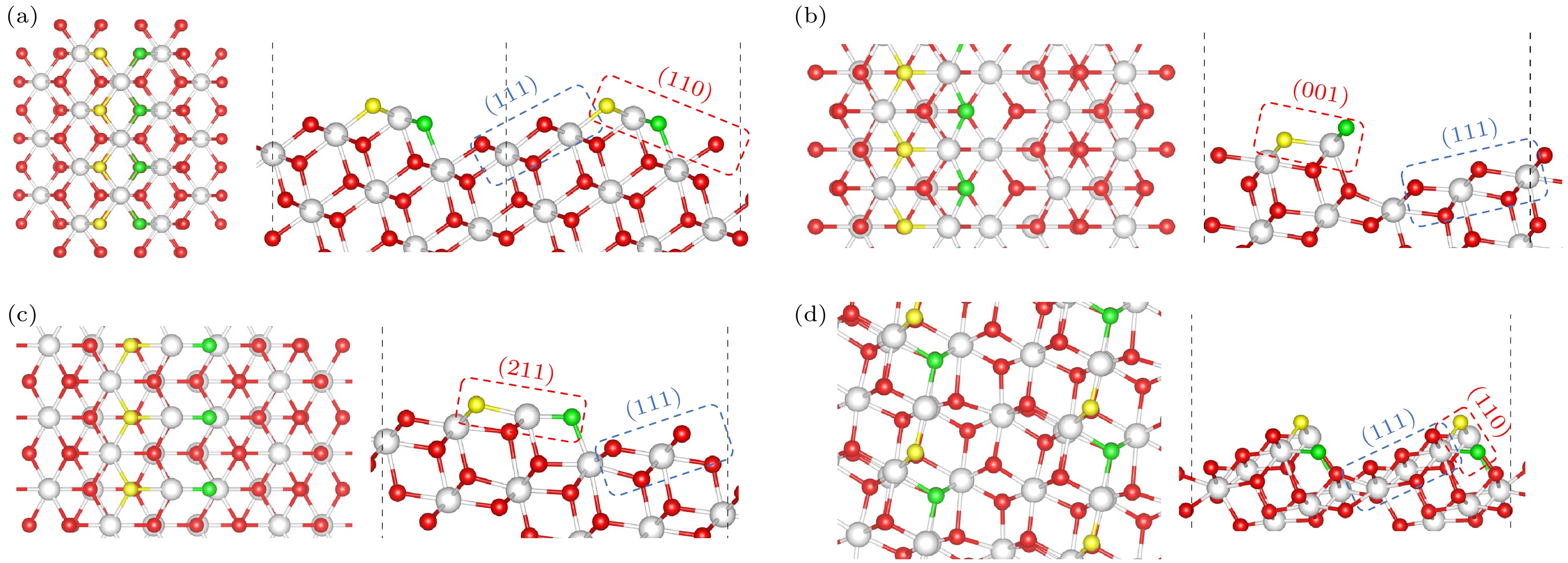
图 5 (a) I型台阶, (b) II型台阶, (c) II*型台阶, (d) III型台阶构建的化学计量CeO2表面. 台阶上边缘的氧(OT)和靠近台阶下边缘的氧(OE)分别用黄色和绿色代表(左: 俯视图; 右: 侧视图)
Fig. 5. Calculated structures of stoichiometric CeO2 vicinal surfaces built for (a) Type I steps, (b) Type II steps, (c) Type II* steps, (d) Type III steps. The oxygen at the border of the upper (111) terrace (OT) and the oxygen at the edge close to the lower (111) terrace (OE) are highlighted in yellow and green, respectively (Left: Top view; Right: Side view).
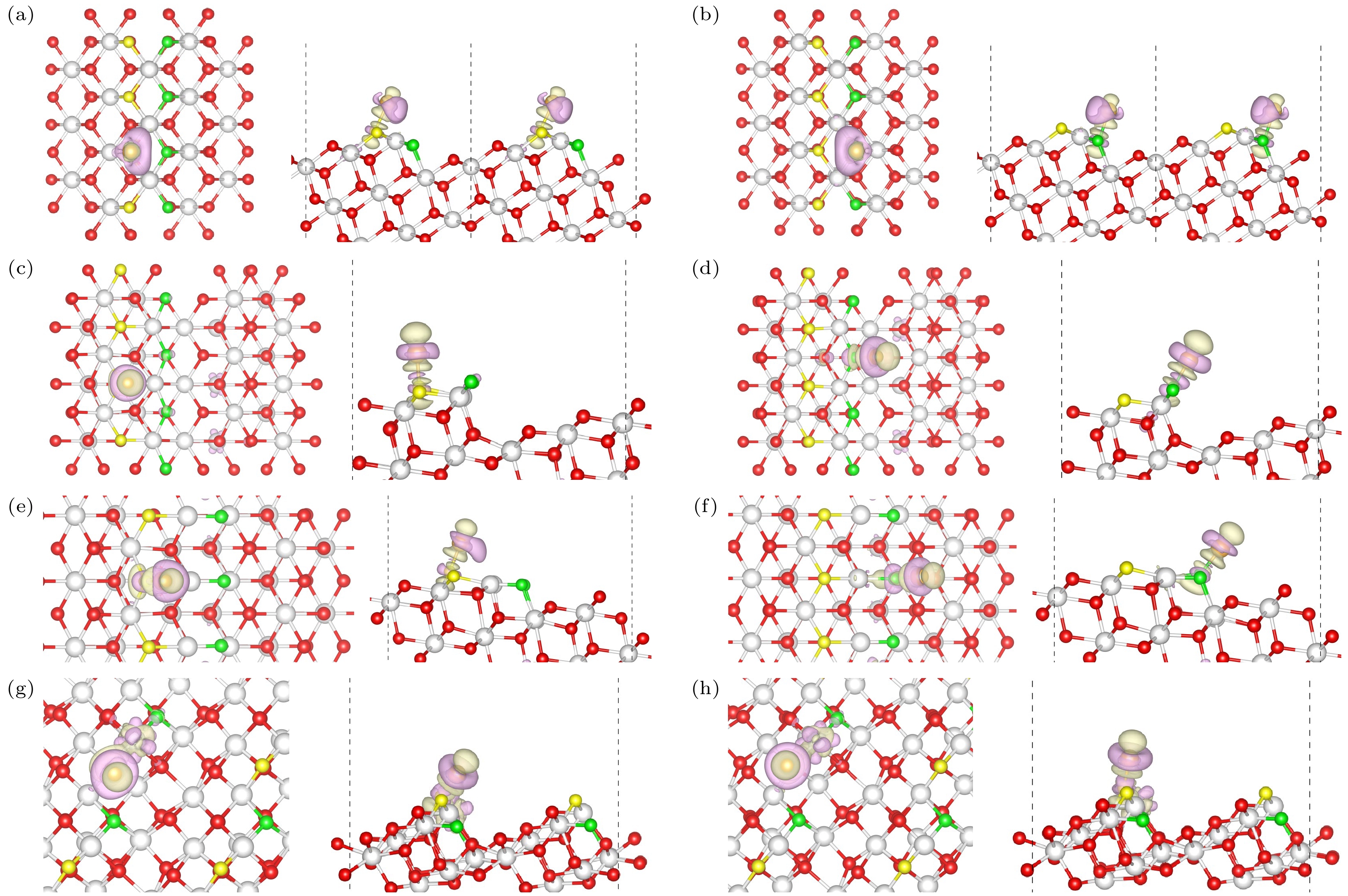
图 6 Au/CeO2(111)台阶上的差分电荷密度(左: 俯视图; 右: 侧视图) (a) Type I OT位点, (b) Type I OE位点; (c) Type II OT位点, (d) Type II OE位点; (e) Type II* OT位点, (f) Type II* OE位点; (g) Type III OT位点, (h) Type III OE位点; 图中紫色部分表示该位置电荷减少, 绿色部分表示电荷的增加
Fig. 6. Differential charge density on stepped Au/CeO2(111) surfaces (Left: Top view; Right: Side view): (a) OT of Type I, (b) OE of Type I; (c) OT of Type II, (d) OE of Type II; (e) OT of Type II*, (f) OE of Type II*; (g) OT of Type III, (h) OE of Type III; the purple regions indicate charge depletion, while the green regions indicate charge accumulation at those locations.
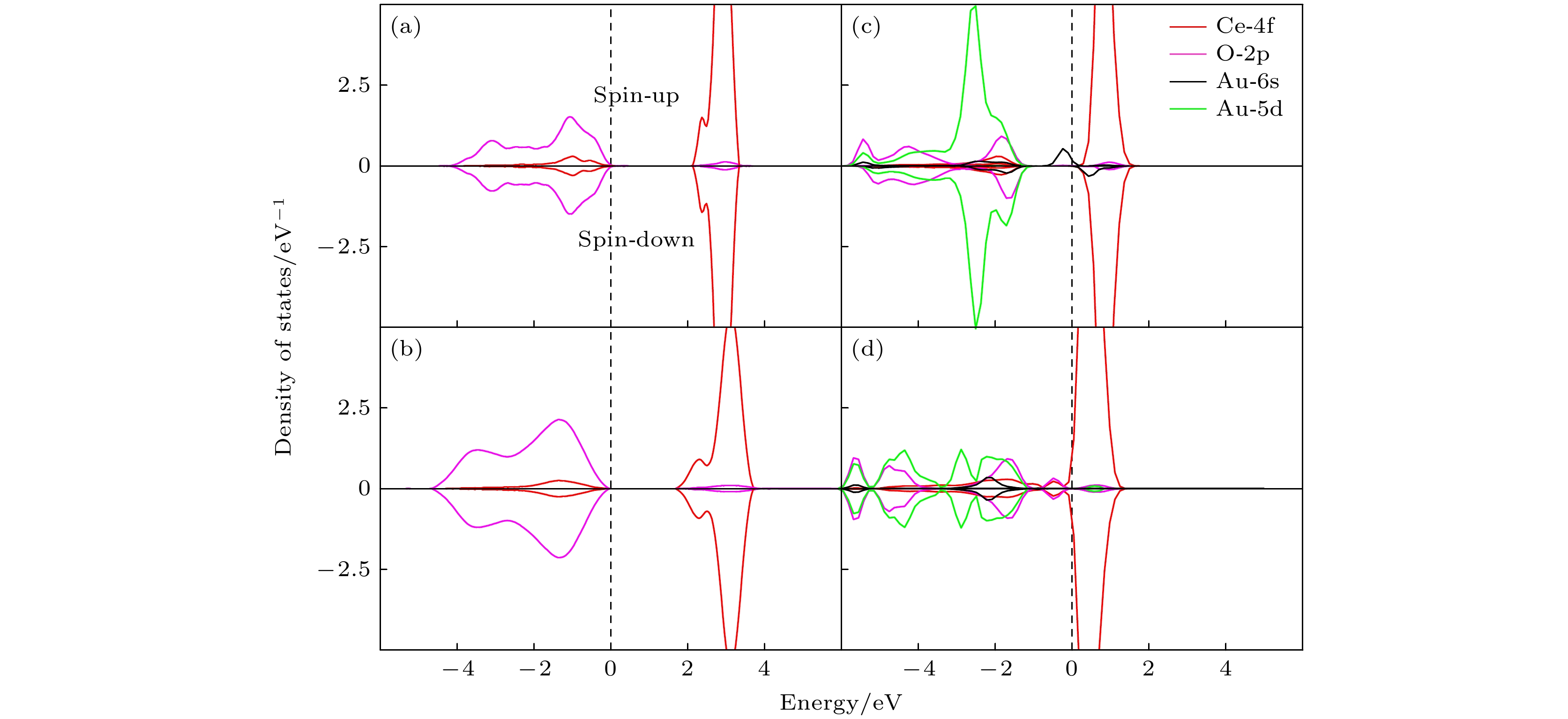
图 7 CeO2(111)表面上的 (a) Type II*, (b) Type III; (c) Au吸附Type II* OE位点; (d) Au吸附Type III OT位点DOS. spin-up 和 spin-down 分别代表自旋向上和自旋向下的电子态
Fig. 7. DOS for (a) Type II* on CeO2 (111) surface, (b) Type III on CeO2 (111) surface; (c) Au adsorbed at OE site of Type II*; (d) Au adsorbed at OT site of Type III; spin-up and spin-down represent the electronic states with spin directed upwards and downwards, respectively.
表 1 Au/CeO2(111)表面吸附构型的能量和几何性质, Eads是吸附能, d[Au-O]和d[Au-Ce]是附着原子到表面原子的距离, d[Ce-O]是铈原子到氧原子的距离
Table 1. Energy and geometric properties of adsorption configurations on the Au/CeO2(111) surface, Eads is the adsorption energy, d[Au–O] and d[Au–Ce] are distances from the adsorbed atoms to surface atoms, and d[Ce-O] is the distance between the cerium atom and the oxygen atom.
Site Eads/eV $ d_\text{[Au-O]}$/Å $ d_\text{[Au-Ce]} $/Å $ d_\text{[Ce-O]} $/Å Ce 0.39 2×3.15
1×3.101×2.99
2×4.86
1×4.913×2.36
3×2.39Od 0.61 1×2.73
1×2.75
1×2.791×3.26
1×3.28
1×3.29
1×5.099×2.38 Ou-Ce 0.77 2.15 1×3.17
1×3.98
1×4.01
1×4.971×2.34
1×2.35
3×2.36
1×2.37Ou 0.95 1.97 1×3.68
1×3.70
1×4.07
1×5.631×2.38
2×2.42
2×2.45
1×2.53Ou-Ou 1.19 2×2.14 1×2.84
2×3.31
1×4.611×2.41
2×2.42
1×2.46
2×2.55 下载: 导出CSV
下载: 导出CSV
表 2 Au/CeO2(111)台阶吸附构型的能量和几何性质, Eads是吸附能, d[Au-O] 和d[Au-Ce]是附着原子到表面原子的距离, d[Ce-O]是铈原子到氧原子的距离, 及不同配位数O和Ce原子
Table 2. Energy and geometric properties of adsorption configurations on stepped Au/CeO2(111) surfaces. Eads is the adsorption energy, d[Au-O] and d[Au-Ce] are the distances from the adsorbed atom to the surface atoms, d[Ce-O] is the distance between the cerium atom and the oxygen atom, along with the coordination numbers of different O and Ce atoms.
Step type Eads/eV $ d_\text{[Au-O]}$/Å $ d_\text{[Au-Ce]}$/Å $ d_\text{[Ce-O]}$/Å Ocoordination Cecoordination OT of TypeⅠ 1.00 1×2.14 1×3.30
1×3.361×2.31
1×2.51
1×2.533 6 OE of TypeⅠ 0.97 1×2.16 1×3.31
1×3.291×2.38
1×2.39
1×2.473 6 OT of TypeⅡ 1.25 1×1.95 1×3.59
2×3.951×2.51
2×2.563 6 OE of TypeⅡ 1.32 1×1.94 1×3.82
1×3.86
1×5.952×2.42 2 6 OT of TypeⅡ* 1.36 1×1.97 1×3.40
1×3.94
1×3.982×2.46 3 5 OE of TypeⅡ* 1.74 1×1.93 1×3.79
1×5.21
1×5.381×2.21
1×2.692 5 OT of Type Ⅲ 2.15 1×1.94 1×3.73
1×3.94
1×4.331×2.42
1×2.472 5 OE of Type Ⅲ 2.14 1×1.93 1×3.79
1×4.00
1×4.791×2.41
1×2.493 5
下载: 导出CSV
-
[1] Haruta M, Kobayashi T, Sano H, Yamada N 1987 Chem. Lett. 16 405
Google Scholar
[2] Li Y J, Wen H, Zhang Q, Adachi Y, Arima E, Kinoshita Y, Nomura H, Ma Z M, Kou L, Tsukuda Y, Naitoh Y, Sugawara Y, Xu R, Cheng Z 2018 Ultramicroscopy 191 51
Google Scholar
[3] Zielinski M, Juszczyk W, Kaszkur Z 2022 RSC Adv. 12 5312
Google Scholar
[4] Berrichi A, Bailiche Z, Bachir R 2022 Res. Chem. Intermed. 48 4119
Google Scholar
[5] Megías-Sayago C, Lolli A, Ivanova S, Albonetti S, Cavani F, Odriozola J A 2019 Catal. Today 333 169
Google Scholar
[6] Li Q L, Xie W, Chen G Q, Li Y F, Huang Y J, Chen X D 2015 Nano Res. 8 3075
Google Scholar
[7] Liu H P, Cao Z L, Yang S Y, Ren Q Y, Dong Z J, Liu W, Li Z A, Chen X, Luo L L 2024 Nano Res. 17 4986
Google Scholar
[8] Kim C, Thompson L 2006 J. Catal. 244 248
Google Scholar
[9] Chen Y, Hu P, Lee M H, Wang H 2008 Surf. Sci. 602 1736
Google Scholar
[10] Hernández N C, Grau-Crespo R, De Leeuw N H, Sanz J Fdez 2009 Phys. Chem. Chem. Phys. 11 5246
Google Scholar
[11] Zhang C, Michaelides A, King D A, Jenkins S J 2010 J. Am. Chem. Soc. 132 2175
Google Scholar
[12] Engel J, Francis S, Roldan A 2019 Phys. Chem. Chem. Phys. 21 19011
Google Scholar
[13] Shu P L, Tian X, Guo Q, Ren X, Zhao B, Wen H F, Tang J, Li Y J, Yasuhiro S, Ma Z M, Liu J 2024 Phys. Scr. 99 105990
Google Scholar
[14] Teng B T, Wu F M, Huang W X, Wen X D, Zhao L H, Luo M F 2012 ChemPhysChem 13 1261
Google Scholar
[15] Lu Y, Quardokus R, Lent C S, Justaud F, Lapinte C, Kandel S A 2010 J. Am. Chem. Soc. 132 13519
Google Scholar
[16] Lustemberg P G, Pan Y, Shaw B J, Grinter D, Pang C, Thornton G, Pérez R, Ganduglia-Pirovano M V, Nilius N 2016 Phys. ReV. Lett. 116 236101
Google Scholar
[17] Fu Q, Saltsburg H, Flytzani-Stephanopoulos M 2003 Science 301 935
Google Scholar
[18] Bezkrovnyi O, Bruix A, Blaumeiser D, Piliai L, Schötz S, Bauer T, Khalakhan I, Skála T, Matvija P, Kraszkiewicz P, Pawlyta M, Vorokhta M, Matolínová I, Libuda J, Neyman K M, Kȩpiński L 2022 Chem. Mater. 34 7916
Google Scholar
[19] Tibiletti D, Amieiro-Fonseca A, Burch R, Chen Y, Fisher J M, Goguet A, Hardacre C, Hu P, Thompsett D 2005 J. Phys. Chem. B 109 22553
Google Scholar
[20] 冯婕, 郭强, 舒鹏丽, 温阳, 温焕飞, 马宗敏, 李艳君, 刘俊, 伊戈尔·弗拉基米罗维奇·雅明斯基 2023 72 110701
Google Scholar
Feng J, Guo Q, Shu P L, Wen Y, Wen H F, Ma Z M, Li Y J, Liu J 2023 Acta Phys. Sin. 72 110701
Google Scholar
[21] Perdew J P, Burke K, Ernzerhof M 1996 Phys. ReV. Lett. 77 3865
Google Scholar
[22] Cococcioni M, De Gironcoli S 2005 Phys. Rev. B 71 035105
Google Scholar
[23] Zheng Z Y, Wang D, Zhang Y, Yang F, Gong X Q 2020 Chin. J. Cata. 41 1360
Google Scholar
[24] Wilson E L, Grau-Crespo R, Pang C L, Cabailh G, Chen Q, Purton J A, Catlow C R A, Brown W A, De Leeuw N H, Thornton G 2008 J. Phys. Chem. C 112 10918
Google Scholar
[25] Ma Z M, Shi Y B, Mu J L, Qu Z, Zhang X M, Li Q, Liu J 2017 Appl. Surf. Sci. 394 472
Google Scholar
[26] Owen C J, Jenkins S J 2021 J. Chem. Phys. 154 164703
Google Scholar
[27] Chen L J, Tang Y, Cui L, Ouyang C, Shi S 2013 J. of Power Sources 234 69
Google Scholar
[28] Kim H Y, Henkelman G 2013 J. Phys. Chem. Lett. 4 216
Google Scholar
[29] Kozlov S M, Neyman K M 2014 Phys. Chem. Chem. Phys. 16 7823
Google Scholar
[30] Olbrich R, Pieper H H, Oelke R, Wilkens H, Wollschläger J, Zoellner M H, Schroeder T, Reichling M 2014 Appl. Phys. Lett. 104 081910
Google Scholar
[31] Barth C, Laffon C, Olbrich R, Ranguis A, Parent Ph, Reichling M 2016 Sci Rep. 6 21165
Google Scholar
[32] Shu P L, Guo Q, Tian X, Wei J, Qu Z, Ren X, Wen H F, Tang J, Li Y J, Sugawara Y, Ma Z M, Liu J 2024 Surf. Interfaces 51 104738
Google Scholar
[33] Kozlov S M, Viñes F, Nilius N, Shaikhutdinov S, Neyman K M 2012 J. Phys. Chem. Lett. 3 1956
Google Scholar
[34] Chu D R, Wang Z Q, Gong X Q 2022 Surf. Sci 722 122096
Google Scholar
[35] 温焕飞, 菅原康弘, 李艳君 2020 69 210701
Google Scholar
Wen H F, Sugawara Y, Li Y J 2020 Acta Phys. Sin. 69 210701
Google Scholar
[36] Zhou H, Wang D, Gong X Q 2020 Phys. Chem. Chem. Phys. 22 7738
Google Scholar
[37] Zhou C Y, Wang D, Gong X Q 2019 Phys. Chem. Chem. Phys. 21 19987
Google Scholar
[38] Piliai L, Matvija P, Dinhová T N, Khalakhan I, Skála T, Doležal Z, Bezkrovnyi O, Kepinski L, Vorokhta M, Matolínová I 2022 ACS Appl. Mater. Interfaces 14 56280
Google Scholar
[39] Janssen E M W, Wiegers G A 1978 J. Less-Common Met. 57 47
Google Scholar


 下载:
下载:






计量
- 文章访问数: 4182
- PDF下载量: 113
- 被引次数: 0
